Содержание
- 1 Синдром Тричера Коллинза: причины болезни и лечение заболевания
- 2 Общая информация
- 3 лечение
- 4 Медиа-изображения
- 5 Стадии развития болезни
- 6 Операция
- 7 Продолжительность жизни
- 8 Симптомы
- 9 Синдром Тричера Коллинза причины
- 10 причины
- 11 Диагностика
- 12 Возможные осложнения и прогноз
- 13 Возможные осложнения и прогноз
- 14 Видео о синдроме Тричера Коллинза
- 15 Операция
- 16 Медицинская справка
- 17 Стадии развития болезни
- 18 Ссылки[править | править код]
Синдром Тричера Коллинза: причины болезни и лечение заболевания
Синдромом Тричера-Коллинза называют заболевание наследственного характера, характеризующееся деформацией костных структур черепа и лица. Другое название болезни – челюстно-лицевой дизостоз.
Патология возникает у одного новорожденного на 50 тысяч малышей. Подробнее о синдроме Тричера-Коллинза и жизни людей, имеющих дефекты на уровне генов, рассказано в статье.
Синдром Тричера-Коллинза: причины заболевания
Эдвард Тричер Коллинз описал основные нарушения, возникающие у больных людей, более ста лет назад. Патология получила соответствующее название. Современная медицина предпочитает использовать другие термины – мандибулофасциальный дизостоз или синдром Франческетти-Коллинза.
Основным фактором, который приводит к развитию заболевания, считается врожденная аномалия в гене 5-ой хромосомы.
Этот ген считается ответственным за кодировку белка, принимающего участие в правильном формировании костей лицевой части черепа в период внутриутробного развития ребенка. Белок называется ядрышковым фосфопротеином.
В конце первого месяца беременности лицевой отдел плода заполняется мезенхимальными тканями, которые на протяжении второго месяца уже дифференцируются в кости и соединительнотканные элементы.
Заболевание может возникнуть в случае тератогенного влияния следующих факторов:
- этанол и его производные;
- радиоактивное излучение;
- цитомегаловирус;
- токсоплазмоз;
- влияние гербицидов;
- препараты на основе ретиноевой кислоты;
- противосудорожные лекарства;
- психотропные препараты.
Симптомы патологии
Основными проявлениями синдрома Тричера-Коллинза считаются:
- опущенные углы глаз;
- недоразвитость скуловых костей с обеих сторон;
- отсутствие части тканей века;
- недоразвитость кости челюсти;
- отсутствие или малый размер раковин ушей;
- отсутствие или заращение наружных слуховых проходов врожденного характера;
- анатомические дефекты среднего отдела уха;
- «птичье лицо».
Так как височная кость является сложным элементом черепно-лицевого отдела, в некоторых случаях всю тяжесть поражений анатомо-физиологических особенностей наружного слухового прохода и внутренних структур уха оценить достаточно трудно.
Визуальный осмотр больного человека позволяет определить отсутствие ресниц на нижнем веке, в некоторых случаях – расщелину неба. При синдроме Тричера-Коллинза возможны врожденные дефекты и нарушения работы со стороны сердца и элементов опорно-двигательного аппарата (помимо лицевого отдела).
 Болезнь хрустальная: причины, фото, продолжительность жизни
Болезнь хрустальная: причины, фото, продолжительность жизниОсобенности жизни человека с синдромом Тричера-Коллинза можно увидеть в документальном фильме «Новое лицо Джулианы».
На момент одиннадцати лет ребенок перенес уже более 50 операций, но лечение еще продолжается.
Родители Джулианы имеют также старшую здоровую дочь. Они хотели еще одного ребенка, однако, побоялись его планировать из-за возможности передачи генной мутации. Взамен родители удочерили украинскую девочку, имеющую такую же генетическую болезнь, но в менее тяжелом проявлении.
Лечение синдрома Тричера-Коллинза
Это заболевание, как и множество других генетических дефектов и нарушений, лечению не поддается. Используется паллиативная терапия, которая позволяет поддержать жизнь больного на достаточном уровне.
Для коррекции слуховых функций используют слуховые аппараты, а чтобы улучшить речь, необходима помощь специалиста-логопеда.
Показан ряд и других пластических операций для лечения и восстановления анатомических дефектов.
Джон Ланкастер и синдром Тричера-Коллинза
Джон Ланкастер – парень, проживающий в Великобритании. На данный момент ему 26 лет. Биологические родители отказались от Джона сразу после его рождения, они не смогли смириться с грозным заболеванием, однако ребенка усыновила другая пара, которая смогла обеспечить его всем необходимым.
Ланкастер перенес несколько пластических операций, которые немного изменили его внешность.
На данный момент он показывает своим примером, что даже с генетическими аномалиями можно жить и радоваться жизни.
 Миастения: продолжительность жизни и прогноз
Миастения: продолжительность жизни и прогнозСестра Аделины Сотниковой и синдром Тричера-Коллинза
Аделина Сотникова – российская олимпийская чемпионка по фигурному катанию. Ее младшая сестра Маша имеет генетическое заболевание, о чем стало известно недавно, после победы Аделины в Сочи.
После рождения младшей сестры родителей Аделины стали готовить в роддоме к тому, что больного синдромом Тричера-Коллинза ребенка придется оставить, но они отказались, приняв решение заботиться о дочери и бороться за ее здоровье.
Свои гонорары Аделина Сотникова тратила на лечение сестры Маши. Девушке еще предстоят хирургические вмешательства, поскольку она продолжает расти.
Общая информация
Синдром Тричера Коллинза, фото которого можно увидеть в этой статье, – это генетическое заболевание, при котором происходит мутация гена 5-й хромосомы, отвечающего за формирование костей лицевой части черепа. Кроме этого, могут быть и другие нарушения.
При данном заболевании прекращается нормальный процесс синтеза и удвоения ДНК. Это происходит на 3-4 неделе после оплодотворения. В результате уже на втором месяце беременности в костях лица формируются участки из соединительной ткани, так как половины генетического материала становится недостаточно для нормального развития организма.
Деление клеток в структурах правой жаберной дуги зародыша нарушается.
Болезнь передается по наследству и может наблюдаться у членов одной семьи в 2-3 поколениях. Она имеет различный характер и степень тяжести, что затрудняет генетическое консультирование (профилактику наследственных болезней). Распространенность составляет 1 случай на 50 тыс. новорожденных.
Измененная копия гена является доминирующей, поэтому риск рождения ребенка с этой патологией высок, даже если болен только один из родителей (около 90% вероятности). В случае, когда данным заболеванием страдают оба родителя, то такая вероятность составляет 100%.
В медицине существуют и другие названия синдрома Тричера Коллинза:
 Спондилез поясничного отдела позвоночника 2 степени прогноз жизни
Спондилез поясничного отдела позвоночника 2 степени прогноз жизни- синдром Франческетти;
- мандибулофациальный дизостоз;
- синдром Томпсона.
лечение
Лечение людей с TCS может включать вмешательство профессионалов из разных дисциплин. В первую очередь заботятся о дыхании и кормлении из-за гипоплазии нижней челюсти и обструкции гортани языком. Иногда им может потребоваться трахеостомия для поддержания проходимости дыхательных путей и гастростомия для обеспечения адекватного потребления калорий и защиты дыхательных путей. Корректирующая хирургия лица проводится в определенном возрасте, в зависимости от состояния развития.
Обзор настоящих рекомендаций:
- Если присутствует волчья пасть , лечение обычно проводится в возрасте 9–12 месяцев. Перед операцией необходима полисомнография с установленной небной пластиной. Это может предсказать послеоперационную ситуацию и дает представление о вероятности наличия апноэ во сне (СОАС) после операции.
- Потеря слуха лечится усилением костной проводимости, логопедом и образовательными вмешательствами, чтобы избежать языковых / речевых проблем. Кости привязанного слуховой аппарат является альтернативой для людей с уха аномалий.
- Скуловая и орбитальная реконструкция выполняется, когда краниоорбитозигоматическая кость полностью сформирована, обычно в возрасте 5–7 лет. У детей чаще всего используют аутотрансплантат. В сочетании с этой трансплантацией можно использовать липофилинг в периорбитальной области для получения оптимального результата реконструкции. Реконструкция нижнего века колобомой включает в себя использование мышечно — кожный лоскут, который поднимается , и таким образом , закрывает дефект веко.
- Реконструкция наружного уха обычно выполняется, когда человеку не менее восьми лет. Иногда можно лечить наружный слуховой проход или среднее ухо.
- Оптимальный возраст для челюстно-нижнечелюстной реконструкции остается спорным; с 2004 г. использовалась эта классификация:
- Тип I (легкий) и Тип IIa (умеренный) 13–16 лет
- Тип IIb (порок развития от умеренной до тяжелой) при зрелости скелета
- Тип III (тяжелый) 6–10 лет
- Когда зубы прорезаются, зубы должны находиться под наблюдением ортодонта, чтобы убедиться в отсутствии аномалий. При обнаружении таких аномалий, как вывих или чрезмерный рост зубов, необходимо как можно скорее принять соответствующие меры.
- Ортогнатическое лечение обычно проводится после 16 лет; к этому моменту все зубы встали на свои места, а челюсти и протезы стали зрелыми. При обнаружении СОАС уровень обструкции определяется с помощью эндоскопии верхних дыхательных путей. Продвижение нижней челюсти может быть эффективным способом улучшить как дыхание, так и эстетику, в то время как пластика подбородка только восстанавливает профиль.
- Если необходима реконструкция носа, ее обычно проводят после ортогнатической хирургии и после 18 лет.
- Контур мягких тканей лица обычно требует коррекции в более позднем возрасте из-за зрелости лицевого скелета. Использование микрохирургических методов, таких как свободный перенос лоскута , улучшило коррекцию контуров мягких тканей лица. Еще один метод улучшения контуров мягких тканей лица — липофилинг. Например, липофилинг используется для реконструкции век.
Потеря слуха
Потеря слуха при синдроме Тричера Коллинза вызвана деформированными структурами внешнего и среднего уха. Потеря слуха обычно двусторонняя с кондуктивной потерей около 50-70 дБ. Даже в случаях с нормальными ушными раковинами и открытыми наружными слуховыми проходами цепочка слуховых косточек часто деформируется.
Попытки хирургической реконструкции наружного слухового прохода и улучшения слуха у детей с СКС не дали положительных результатов.
Слуховая реабилитация с использованием слуховых аппаратов с костной фиксацией (BAHA) или обычных аппаратов костной проводимости оказалась предпочтительнее хирургической реконструкции.
Психиатрическая
Расстройство может быть связано с рядом психологических симптомов, включая тревогу, депрессию, социальную фобию и беспокойство по поводу образа тела; люди также могут сталкиваться с дискриминацией, издевательствами и обзываниями, особенно в молодом возрасте. Эти вопросы должны учитывать многопрофильная команда и родительская поддержка.
Медиа-изображения
Статья в New York Times от июля 1977 года, которая в последующие недели была перепечатана во многих газетах по всей стране, впервые привлекла внимание многих людей к этой болезни.
Беспорядок был показан в шоу Nip / Tuck в эпизоде » «.
В фильме TLC « Born Without a Face» изображена Джулиана Ветмор, которая родилась с самым тяжелым случаем в истории болезни этого синдрома, и у нее отсутствуют 30-40% костей на лице.
В 2010 году документальный фильм BBC Three « Люби меня, люби мое лицо» рассказывал о случае человека, Джоно Ланкастера, с этим заболеванием. В 2011 году BBC Three вернулись к Джоно, чтобы осветить его и его партнершу Лору стремление создать семью в фильме « Что, если мой ребенок родился, как я?». , который впервые вышел в эфир в рамках трех сезонов программ BBC о воспитании детей. Первый фильм был показан на BBC One незадолго до первой трансляции второго фильма на BBC Three. Третий фильм Ланкастера BBC Three «В поисках семьи на Facebook» , посвященный усыновлению , вышел в эфир в 2011 году.
В Wonder , то детский роман , главный герой является ребенок , который имеет синдром Treacher Collins. В ноябре 2017 года вышла экранизация 2017 года с Джулией Робертс , Оуэном Уилсоном и Джейкобом Трембле в главных ролях .
Элисон Мидстокк , актриса и модель с этим заболеванием.
Стадии развития болезни
Операция
Аномальное развитие лицевых костей помогает устранить челюстно-лицевая и пластическая хирургия. В сложных случаях требуется проведение целой серии таких операций.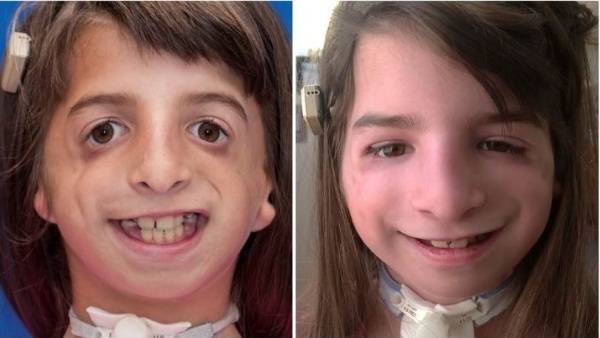
Оперативной корректировке поддаются следующие нарушения:
- дефекты ушной раковины и среднего уха;
- искаженный овал лица (липоскульптура, вытяжение, костные трансплантаты);
- «волчья пасть»;
- расщелины неба;
- дефекты век;
- ограниченное открытие рта.
Этиологического лечения для синдрома Тричера Коллинза не существует. Как показывают фото, степень выраженности этого генетического отклонения может быть различной. Для пациентов применяется терапия, поддерживающая их жизнь на достаточном уровне.
Наиболее распространенным осложнением является ухудшение слуха, для устранения которого используются слуховые аппараты. С целью восстановления анатомических дефектов по возможности проводят пластическую хирургию.
Оформление статьи: Владимир Великий
Продолжительность жизни
Симптомы
Такой необычный недуг имеет много самых разных проявлений. Причём у одного человека с этим диагнозом могут наблюдаться далеко не все возможные дефекты. А так как это заболевание врождённое, то первые признаки болезни можно наблюдать сразу после рождения ребёнка.
Основные проявления болезни – это многочисленные уродства лица. При этом чаще всего наблюдается неправильное формирование глазной щели. В таком случае наружный угол глаза всегда направлен не вверх, как бывает обычно, а вниз. Такое явление наблюдается с обеих сторон. Веки при этом имеют форму треугольника, что носит название колобома.
Второй важный диагностический признак – это недоразвитие скуловой кости. Кости скул очень маленькие, что в свою очередь приводит к неправильной симметрии лица. Нижняя челюсть также имеет некоторое недоразвитие и, как правило, очень маленькая. При этом наблюдается большой рот.
Недоразвитие касается и зубов. В некоторых случаях они могут и вовсе отсутствовать на протяжении всей жизни, но чаще всего зубы сильно расставлены друг от друга, что формирует неправильный прикус.
Третий немаловажный признак – это или полное отсутствие, или недоразвитие ушных раковин и слухового прохода. Из-за этого дети не могут иметь нормальный слух.
Это заболевание имеет несколько стадий в своём развитии. При начальной стадии изменения на лице практически не заметны. При средней степени тяжести, что выявляется чаще всего, отмечаются вышеперечисленные нарушения. При тяжёлой степени у ребёнка практически невозможно рассмотреть черты лица.
Видео: Девочка без лица. История джулианы уэтмор
Синдром Тричера Коллинза причины
Синдром Тричера Коллинза является аутосомно — доминантным заболеванием. Это значит, что дефектный ген этой болезни не связан с половой хромосомой, а значит, равносильно может появиться и у мужчин и у женщин. Кроме того, этот ген является доминантным, что означает, что при его наличии в организме, синдром Тричера Коллинза проявится в 100% случаев наличия этого гена.
Таким образом, развитие заболевания Тричера Коллинза не зависит от воздействия каких-либо вредных внешних и внутренних факторов. Можно сказать, что это заболевание уже заложено в генетический код будущего ребенка и начинает раскрываться задолго до его рождения.
Причина появления синдрома Тричера Коллинза у новорожденного ребенка кроется еще на этапе эмбриогенеза плода и процесса закладки органов. Именно в это время, а точнее на 7-й неделе развития эмбриона, происходит мутация в определенной хромосоме человеческого генетического кода – 5-й хромосоме. Эта хромосома является самой длинной в геноме человека, и именно она отвечает за синтез материала для будущего костного скелета. В результате в этой хромосоме происходит особая мутация — так называемая «нонсенс-мутация». Особенность этой мутации кроется в особенностях внутриклеточного синтеза белка. В норме такой важный процесс как биосинтез белка происходит следующим образом: цепь ДНК «перезаписывает» информацию на вспомогательную единицу – РНК. Буквально говоря РНК клонируется, записывая на себя определенную последовательность участков ДНК. Эти участки представляют собой определенную последовательность составных частей нуклеотидов, каждый из которых несет свою особую информацию. Но кроме нуклеотидов существуют и особые гены, которые получили название «стоп-кодонов». Эти гены выполняют особую функцию — в последующей сборке белка на основе РНК они в заканчивают постройку молекулы белка.
Народ опешил! Суставы восстановятся за 3 дня! Приложите…
Мало кто знает, но именно это лечит суставы за 7 дней!
После того, как будет создана РНК с полной информацией, аналогичной информации материнской ДНК, она транспортируется в особый клеточный орган-рибосому. Именно она занимается синтезированием будущего белка — основы клеточной структуры определенных органов. РНК проходит сквозь рибосому, а точнее через ее функциональные участки. Эти участки взаимодействуют с РНК, «считывают» с нее информацию и вырабатывают белковые цепи, где каждая из них соответствует своему нуклеотиду на РНК. Но когда функциональный центр взаимодействует с описанным выше геном стоп кодоном, то он получает информацию о прекращении синтеза белка. Говоря своими словами, стоп кодоны, как бы «отрезают» от общей массы отдельные полипептидные цепи, где каждая имеет свою структуру. Позже эти цепи будут собраны в белковые молекулы.
Но при синдроме Тричера Коллинза в 5-й хромосоме, в ее гене под названием TCOF1 происходит сбой — на месте нормальных нуклеотидов, способных создать полипептидную цепь, образуется стоп-кодон. В результате, при дальнейшем синтезе происходит преждевременное окончание сборки белка, и такой белок получается дефектным. Как результат, развивается синдром гаплонедостаточности — количества образуемого белка при такой недостаточности просто недостаточно для синтезирования будущего прототипа костной структуры лицевой части черепа и последующего правильного развития из него самой костной структуры.
Как следствие этого, развивается формирование целого ряда деформаций лицевой части черепа: нарушение пропорций ее костной части, атрезия (недоразвитие) ушных раковин и наружного слухового прохода, полное или частичное нарушение формирования правильных черт лица.
причины
Причиной синдрома Тричера-Коллинза является мутация (то есть аномалия в нормальной последовательности ДНК) одного из человеческих генов, известных как TCOF1, POLR1C и POLR1D .
Другими словами, человек страдает от синдрома Тричера-Коллинза, когда один из генов TCOF1, POLR1C и POLR1D имеет аномалию в последовательности ДНК.
Гены, вовлеченные в синдром Тричера Коллинза: сайт и нормальная функция
Предпосылка: гены, присутствующие в хромосомах человека, представляют собой последовательности ДНК, задачей которых является производство фундаментальных белков в биологических процессах, необходимых для жизни, включая рост и репликацию клеток.
Гены TCOF1, POLR1C и POLR1D находятся соответственно в хромосоме 5, хромосоме 6 и 13 хромосоме .
В отсутствие мутаций против них (когда у человека, не затронутого синдромом Трэчера Коллинза), каждый из вышеупомянутых генов продуцирует белок, играющий ключевую роль в правильном развитии костей и мягких тканей лица; более конкретно, они продуцируют белки, ответственные за выработку рибосомальной РНК (особого типа РНК), которая служит для регулирования процессов роста костей и мягких тканей лица.
Что вызывает мутацию генов синдрома Тричера Коллинза?
При наличии мутаций, ответственных за синдром Тришера Коллинза, гены TCOF1, POLR1C и POLR1D теряют способность экспрессировать функциональные белки, что приводит к отсутствию направляющих элементов во время процессов развития костей и мягких тканей на лице.
Другими словами, генетические мутации в происхождении синдрома Treacher Collins вызывают отсутствие агентов, ответственных за регулирование правильного роста костных тканей и мягких тканей, которые составляют человеческое лицо.
Наследственное или приобретенное заболевание?
Синдром Тричера-Коллинза может быть результатом наследственной мутации (т.е. передаваемой через родительские средства) или спонтанно приобретенной мутации, возникшей из ничего и без точных причин во время эмбрионального развития (т.е. после оплодотворения сперматозоида). яйцо и эмбриогенез начался).
Статистически говоря, синдром Тричера-Коллинза чаще является приобретенным состоянием, чем унаследованным состоянием; фактически, это связано с мутацией приобретенного типа в 60% клинических случаев и с наследственной мутацией в оставшихся 40% (*)
* Примечание: рассматриваемые статистические данные относятся исключительно к случаям синдрома Тричера-Коллинза из-за мутации генов TCOF1 и POLR1D.
Для клинических случаев, связанных с мутацией гена POLR1C, нет информации о типе мутации.
Чтобы понять …
- Каждый человеческий ген представлен в двух экземплярах, называемых аллелями, материнского и отцовского происхождения.
- Генетическое заболевание является аутосомно-доминантным, когда мутация только одной копии гена, вызывающая его, достаточна для проявления.
- Наследственное заболевание является аутосомно-рецессивным, когда необходима мутация обеих копий гена, вызывающая его.
Синдром Тричера-Коллинза из-за мутации генов TCOF1 и POLR1D обладает всеми характеристиками аутосомно-доминантного заболевания ; напротив, синдром Тричера-Коллинза, связанный с мутацией гена POLR1C, имеет генетические характеристики, типичные для аутосомно-рецессивного заболевания .
Диагностика
Диагноз СТК ставится на основании тщательной клинической оценки, подробного анамнеза пациента и определения характерных физических признаков. При рождении присутствуют многие сопутствующие аномалии, такие как пороки развития или отсутствие наружного уха.
— Клиническое тестирование и обследование.
Специализированные рентгенологические исследования подтвердят наличие и/или степень определенных наблюдаемых черепно-лицевых аномалий. Например, такие визуальные исследования показывают аномальную малость челюсти (микрогнатия) из-за недоразвития кости нижней челюсти (гипоплазия нижней челюсти), наличие и/или степень гипоплазии, затрагивающей определенные части черепа, и/или наличие дополнительных пороков развития уха, которые нельзя увидеть во время клинической оценки.
Кроме того, у тех больных, у которых наблюдается мало признаков, тщательное клиническое обследование и рентгенография черепно-лицевой области могут продемонстрировать едва различимое присутствие определенных характерных признаков (например, гипоплазию скуловых дуг), связанных с синдромом. Поскольку синдром Тричера-Коллинза обладает несколькими физическими особенностями, которые могут встречаться при других черепно-лицевых синдромах, многие исследователи рекомендуют делать диагностическое подтверждение посредством молекулярно-генетического тестирования и/или, в некоторых случаях, тщательной, детальной семейной истории.
Молекулярно-генетическое тестирование для подтверждения диагноза доступно в коммерческих и академических исследовательских лабораториях для выявления мутаций в генах TCOF1, POLR1B, POLR1C и POLR1D. Приблизительно 80% людей имеют идентифицируемую мутацию гена TCOF1. Кроме того, генетическое подтверждение TCOF1, POLR1B, POLR1C или POLR1D мутаций может быть обнаружена до рождения (пренатально) путем амниоцентеза и взятия проб ворсинчатого хориона, если мутация была выявлена у больного члена семьи. В некоторых случаях ультразвуковое исследование плода, в котором используются отраженные звуковые волны для создания изображения развивающегося плода, может выявить характерные признаки, указывающие на наличие СТК у ребенка. Родственники, особенно родители и братья или сестры, лиц с диагнозом СТК должны быть тщательно обследованы, поскольку легкие случаи часто остаются нераспознанными и не диагностируемыми.
Возможные осложнения и прогноз
Исход недуга зависит от степени выраженности патологических изменений. Как правило, костные деформации не несут угрозы жизни и здоровью ребенка. Малыши не отстают в умственном развитии, однако испытывают трудности с социальной адаптацией, как из-за специфической внешности, так и в результате нарушений слуха и речи.
Опасными могут быть сочетающиеся с синдромом Фраческетти пороки сердца, а также дефекты костей, приводящие к затруднению дыхания пациента. В таких случаях прогноз более осторожный. Большинство проблем, возникающих на фоне этой генетической мутации, поддаются хирургическому лечению.
Специфических методов предупреждения развития синдрома Тричера Коллинза не разработано. Эта особенность связана с тем, что контролировать хромосомные мутации не удается. Поэтому профилактика возникновения дефектов направлена на правильное планирование беременности. Оно подразумевает проведение кариотипирования родителей с целью выявления хромосомных аномалий. Однако даже такой подход не дает гарантий отсутствия заболевания у будущего ребенка. Это связано с широкой распространенностью спонтанных мутаций, приводящих к формированию дизостоза.
* Факторы синдрома Трейчера Коллинза, под редакцией: Чарльз Патрик Дэвис, MD, PhD
- Синдром Трехара Коллинза (TCS) — это состояние (генетическое заболевание), которое изменяет развитие костей и других тканей в лице.
- Признаки и симптомы варьируются от почти незаметных изменений лица до тяжелых изменений лица и уха, расщелины неба и ограниченных дыхательных путей
- Характеристики TCS включают черепно-лицевые или мандибулофациальные аномалии:
- Глаза, которые уклоняются от носа
- Очень немного ресниц и выемки в нижних веках (глаз coloboma)
- Уши, которые отсутствуют или необычно сформированы
- У некоторых людей может быть потеря слуха
- Маленькая челюсть
- Ребенок с ТКС может иметь апноэ во сне и / или проводящую потерю слуха; для потери слуховой функции может потребоваться ресурс для предоставления слуховых аппаратов для детей.
- Некоторые люди могут серьезно пострадать, и у них могут развиться опасные для жизни проблемы с дыханием (инфантильное апноэ).
- Другие аномалии могут затруднить дыхание и кормление ребенка из-за суженной обструкции носовых дыхательных путей.
- У ребенка могут быть особенности «последовательности Пьера Робена», в которой язык находится дальше в горле, чем обычно (глоссоптоз), с или без, и неполное расщепление неба рта и обструкции дыхательных путей.
Возможные осложнения и прогноз
Если заболевание не сопровождается грубыми пороками развития внутренних органов, то прогноз для жизни и здоровья ребенка в будущем благоприятен. При наличии тугоухости снижается способность к развитию речи, письма, обучению.
Необычная внешность также мешает получению навыков социального общения, так как другие дети стараются избегать сверстников с врожденными аномалиями и уродствами. В результате у ребенка может быть значительно снижена самооценка, что требует консультации со стороны психолога. Проведение ряда пластических операций позволяет частично сгладить дефекты развития.
Тугоухость часто ошибочно диагностируется как недоразвитие психики. Умственная отсталость у таких детей встречается редко, а некоторые имеют способности выше среднего
Поэтому у таких пациентов важно на ранней стадии выявлять нарушения слуха и корректировать их
Врачи рекомендуют это делать в обязательном порядке до достижения ребенком шестимесячного возраста. С 3-х месяцев он может носить аппарат для костного проведения звуковых волн, а после 3 лет возможна установка имплантата за ушами. Также очень большое значение имеют занятия с сурдопедагогом.
В первые месяцы жизни недоразвитость нижней челюсти может способствовать выпадению языка и перекрытию дыхательных путей, что является потенциально опасным для жизни ребенка. В последующем могут возникнуть затруднения в приеме пищи из-за ограниченной возможности открытия рта (в различной степени тяжести).
Видео о синдроме Тричера Коллинза
Операция
Медицинская справка
Стадии развития болезни
Синдром Тричера Коллинза, фото которого приведено выше, условно делят на 3 стадии:
- Начальный этап, когда у новорожденного наблюдается незначительное деформирование лицевой части черепа.
- 2-я стадия, на которой происходят нарушение проходимости дыхательных путей, глотания, зрения и слуха.
- 3-я стадия, которая характеризуется грубыми деформациями черепа из-за неравномерного роста костей в течение многих лет.
Первые признаки заболевания можно заметить сразу после рождения ребенка. Их выраженность может быть различной – от едва заметных отклонений до очень тяжелых форм. Анатомические аномалии без хирургического вмешательства постепенно прогрессируют по мере роста и развития ребенка.

 Правила жизни с подагрой. первая помощь при приступе
Правила жизни с подагрой. первая помощь при приступе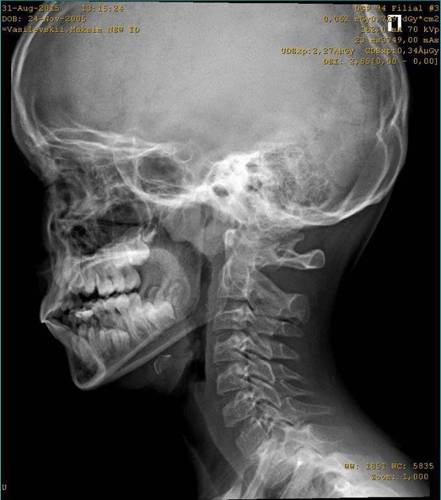 Что такое аномалия киммерли, симптомы, продолжительность жизни и как лечить болезнь?
Что такое аномалия киммерли, симптомы, продолжительность жизни и как лечить болезнь? Покраснение кожи лица
Покраснение кожи лица Хедшот: история шутеров от первого лица
Хедшот: история шутеров от первого лица