Содержание
- 1 Содержание
- 2 Классификация
- 3 Синдром Моркио
- 4 Историческая справка
- 5 Симптомы Мукополисахаридоза типа I-S (болезни Шейе; позднего синдрома Гурлера):
- 6 Клиническая картина
- 7 Диагностика
- 8 Историческая справка
- 9 Особенности заболевания
- 10 Литература
- 11 Клиническая картина
- 12 Ловля сига в озере
- 13 Диагноз
- 14 Историческая справка
- 15 Признаки и симптомы
- 16 Ссылки
- 17 Акции и специальные предложения
- 18 Симптомы
- 19 Наследование
- 20 Лечение Мукополисахаридоза типа I-S (болезни Шейе; позднего синдрома Гурлера):
- 21 Клиническая картина
- 22 Что такое мукополисахаридоз?
- 23 Патологическая анатомия
Содержание
Классификация
Синдром Моркио
Синдром Моркио — наследственное заболевание, относящееся к группе мукополисахаридозов и встречающееся преимущественно у мужчин. У больных отмечается непропорциональное отставание в росте: туловище короткое, в связи с чем конечности кажутся длинными. С возрастом уменьшение роста выражено наиболее ярко. Карликовость зависит прежде всего от уплощения тел позвонков. Голова большая, черты лица грубые, нос широкий, нижняя челюсть увеличена, рот приоткрыт, зубы большие, редкие. Шея короткая, поэтому кажется, что голова «сидит на плечах». Грудная клетка широкая, грудина выступает вперед, лопатки расположены высоко, выражен кифоз грудного и поясничного отделов позвоночника. Ограничены движения в плечевых и локтевых суставах. Кисти деформированы по типу косорукости. Имеет место вальгусная установка коленных суставов, плоскостопие. Кожа дряблая, снижен слух, зрение ухудшено вследствие помутнения роговицы. Психических расстройств нет. Повышено выделение с мочой кератансульфа-та. При рентгенологическом исследовании обнаруживается языкообразная деформация нижнегрудных и верхнепоясничных позвонков. В тазобедренных суставах вертлужные впадины широкие, плоские, вдавленные в малый таз, шейки бедер широкие, вальгусно искривлены, головки бедер резко уплощены. Наблюдается также фрагментация больших вертелов и головок бедер. Характерны изменения коленных суставов: эпифизы костей, образующих коленный сустав, фрагментированы, имеют прямоугольную форму, по наружной поверхности эпифизов бедренной и большеберцовой костей отмечается наиболее интенсивное уплощение. Кроме того, имеет место фрагментация надколенника. Постепенно нарастает вальгусная деформация коленного сустава вплоть до подвывиха голени кнутри. Фаланги пальцев кистей и пястные кости утолщены и укорочены, ядра окостенения выявляются с запозданием, имеют неправильную зубчатую форму. Эпифизы голени в дистальном отделе скошены, деформирован блок таранной кости. Стопы деформированы аналогично кистям.
Историческая справка
Заболевание, изначально названное болезнь Пфаундлера — Гурлер, впервые описано двумя педиатрами: австрийским — нем. Hurler Gertrud (1889—1965) и немецким — нем. Pfaundler Meinhard von (1872—1947).
Описанная авторами болезнь проявляется в первые месяцы жизни грубыми чертами лица (гаргоилизм), гепатоспленомегалией, тугоподвижностью суставов, деформацией позвоночного столба. Затем американским офтальмологом Шейе, англ. Н. G. Scheie (1909—1990) описана вторая форма болезни с более поздним началом и более доброкачественным течением, названная синдром Шейе. Позже описана промежуточная форма болезни, названная синдром Гурлер — Шейе.
Эпоним
Заболевание названо в честь одного из первооткрывателей — австрийского педиатраГертруды Гурлер (нем. Gertrud Hurler), (1889—1965).
Симптомы Мукополисахаридоза типа I-S (болезни Шейе; позднего синдрома Гурлера):
Как самая легкая форма МПС-I, синдром Шейе не проявляет яркой клинической картины. Центральная нервная система не страдает. В неврологическом статусе пациент — здоровый человек. Отмечается тугоподвижность суставов, гепатоспленомегалия.
Сердечно-сосудистая система. Поражение сердца наблюдается у большинства больных. Характер поражения аналогичен таковому при мукополисахаридозе IH, однако более типично развитие аортального порока сердца. Аортальная регургитация развивается в школьном возрасте, обуславливает появление левожелудочковой застойной недостаточности. Реже при этом типе мкуополисахаридоза развивается аортальный стеноз. На ЭКГ регистрируется гипертрофия левого желудочка или, наоборот, снижение вольтажа комплексов QRS из-за замещения миокарда нефункционирующей тканью, при этом определяется изменение процесса реполяризации по типу субэндокардиальной ишемии. Рентгенография выявляет чаще умеренную, реже выраженную кардиомегалию. При эхокардиографическом обследовании определяется преимущественное изменение аортальных створок в виде их уплотнения и утолщения, из-за отложения в них мукополисахаридов. Аналогичное поражение визуализируется и в других клапанах, однако не имеющее клинического значения. При аутопсии определяются узелковые изменения и утолщение полулунных и атриовентрикулярных клапанов Хорды митрального клапана короткие и утолщены. просвет коронарных артерий узкий, стенки толстые. При световой микроскопии отложения мукополисахаридных комплексов обнаруживаются в эндокарде, клапанах, медиальном и интимальном слое коронарных сосудов. Поражение магистральных артериальных сосудов менее характерно для данного синдрома. В ряде случаев может отмечаться сужение абдоминальной аорты с развитием артериальной гипертензии.
Клиническая картина
Несмотря на то, что к развитию муколисахаридоза I типа ведёт дефицит одного фермента альфа-L-идуронидазы, заболевание проявляется в трёх клинических вариантах: синдром Гурлер, синдром Шейе и синдром Гурлер — Шейе.
Исторически первым двумя педиатрами: австрийским — нем. Hurler Gertrud (1889–1965) и немецким — нем. Pfaundler Meinhard von (1872–1947) описан синдром, при котором определяется подавляющее большинство компонентов фенотипа, характерных для этой группы наследственных заболеваний, и все они — резко выражены. Описанная авторами болезнь, ставшая прототипом всех мукополисахаридозов I типа, проявляется в первые месяцы жизни грубыми чертами лица (гаргоилизм), гепатоспленомегалией, тугоподвижностью суставов, деформацией позвоночного столба. Позже американским офтальмологом Шейе, англ. Н. G. Scheie (1909–1990) описана вторая форма болезни с более поздним началом и более доброкачественным течением, названная синдром Шейе. Последней описана промежуточная форма болезни, названная синдром Гурлер — Шейе.
Синдром Гурлер
Основная статья: синдром Гурлер
Дети с синдромом Гурлер отличаются низкорослостью (отставание в физическом развитии наблюдается с конца первого года жизни). Характерны признаки гаргоилизма: крупный череп, крутой лоб, запавшая переносица, толстые губы, большой язык, характерное выражение лица («лицо выплевывающего воду»). Кроме того, отмечается короткая шея, ограниченная подвижность суставов (тугоподвижность, главным образом, затрагивает локтевые и межфаланговые суставы пальцев кистей и стоп), фиксированный кифоз на месте перехода грудных позвонков к поясничным, укорочение конечностей, в основном, происходит за счёт проксимальных отделов (бёдер и плеч), в меньшей степени — голеней и предплечий. Весьма своеобразно строение кисти пациента: пальцы короткие, одинаковые по длине (изодактилия) расходятся веерообразно, напоминая трезубец. Нижнепоясничный лордоз способствует выпячиванию живота вперёд, а ягодиц назад. Отмечается гепатоспленомегалия, склонность к формированию пупочной грыжи. Характерно диффузное помутнение роговицы за счёт накопления в ней дерматансульфата. Возможно развитие слабоумия, кариеса зубов, характерной формы ногтевых пластинок в виде часовых стёкол, тугоухость или глухота, низкий хриплый голос, гипертрихоз, сухие и жёсткие волосы. В большинстве случаев в патологический процесс вовлекается сердце — оно увеличивается в размерах, происходят изменения клапанов, миокарда, эндокарда, крупных и коронарных артерий. При рентгенологическом исследовании определяется преждевременное окостенение лямбдовидного шва, расширение турецкого седла, характерная форма позвонков («рыбьи позвонки»), искривление лучевой кости, деформации метафизарных и эпифизарных отделов длинных трубчатых костей, короткие метакарпальные кости и фаланги пальцев. Такие дети обычно не доживают до 10 лет.
 Синдром марфана: фото, симптомы и лечение
Синдром марфана: фото, симптомы и лечение
Синдром Шейе
Основная статья: синдром Шейе
Клиническая симптоматика развивается на фоне нормальной продолжительности жизни, обычно не проявляется до достижения возраста 4 — 5 лет и может включать в себя:
- помутнение роговицы с прогрессирующей потерей зрения вплоть до слепоты;
- гаргоилизм: грубые черты лица, широкий рот с полными губами, прогнатизм;
- избыточный рост волос на теле (гипертрихоз, гирсутизм);
- характерные деформации кистей и стоп;
- тугоподвижность суставов.
Синдром Гурлер — Шейе
Основная статья: синдром Гурлер — Шейе
Синдром Гурлер — Шейе (мукополисахаридоз-I H/S) является менее тяжёлым промежуточным вариантом между синдромами Гурлер и Шейе. Фенотип пациентов также является промежуточным между фенотипом свойственным синдрому Гурлер и Шейе. Существует предположение, что больные синдромом Гурлер — Шейе являются генетическими химерами с одним аллелем синдрома Гурлер и вторым — синдрома Шейе.
Клиническая симптоматика характеризуется в основном кожными проявлениями, сочетающимися с умеренной умственной отсталостью и помутнением роговицы.
Диагностика
В процессе диагностики и дифференциальной диагностики синдрома Гурлер — Шейе определяют не только тип экскретируемого с мочой мукополисахарида (дерматансульфат, гепарансульфат), но и определяют активность специфического фермента (альфа-L-идуронидазы) в различных тканях организма, в том числе и культуре фибробластов кожи.
Историческая справка
Впервые заболевание, изначально названное болезнь Пфаундлера — Гурлер, описано двумя педиатрами: австрийским — (нем. Hurler Gertrud , 1889—1965) и немецким — (нем. Pfaundler Meinhard von , 1872—1947).
Описанная авторами болезнь проявляется в первые месяцы жизни грубыми чертами лица (гаргоилизм), гепатоспленомегалией, тугоподвижностью суставов, деформацией позвоночного столба. Затем американским офтальмологом Шейе, (англ. Н. G. Scheie, 1909—1990) описана вторая форма болезни с более поздним началом и более доброкачественным течением, названная синдром Шейе. Позже описана промежуточная форма болезни, названная синдром Гурлер — Шейе.
Эпоним
Заболевание названо в честь первооткрывателей — австрийского педиатраГертруды Гурлер (нем. Gertrud Hurler, 1889—1965) и американского офтальмолога Гарольда Шейе, (англ. Harold Glendon Scheie, 1909—1990).
 Судорожный синдром
Судорожный синдромОсобенности заболевания
Мукополисахаридоз 1 типа представляет собой целый ряд генетических отклонений, который появился из-за нарушения обмена веществ, а именно гликозаминогликанов. Поражение касается скелета, тормозит нормальное развитие, затрагивает интеллектуальную сферу.
У больных наблюдается следующие проблемы:
- Выраженное деформирование опорно-двигательного аппарата.
- Характерные черты внешности, кажущиеся иногда неправильными.
- Деформирование ушей, строения челюсти, костей черепа.
- Задержка нормального роста тела.
- Отставание в интеллектуальной сфере, психики.
- Снижение или прекращение нормального движения суставов.
- Поражения внутренних органов или подверженность инфекционным болезням.
- Частое появление грыж из-за дефектов развития.
Зарегистрировано заболевание относительно недавно, всего в прошлом столетии. При начале изучения подразумевалось, что это единичное заболевание, но из-за большого количества отличий, стали выделять по разным типам.
Литература
- Harrison’s Principles of Internal Medicine
- G. Hurler. Uber einen Typ multipler Abartungen, vorwiegend am Skelettsystem. Zeitschrift fUr Kinderheilkunde, Berlin, 1919; 24: 220—234. (англ.)
- M. Pfaundler. Demonstrationen Uber einen Typus kindlicher Dysostose. Jahrbuch fUr Kinderheilkunde und physische Erziehung, Berlin, 1920; 92: 420. (англ.)
- H. G. Scheie, G. W. Hambrick Jr., L. A. Barnes. A newly recognised forme fruste of Hurlers disease (gargoylism). American Journal of Ophthalmology, 1962; 55: 753—769. (англ.)
Клиническая картина
Несмотря на то, что к развитию муколисахаридоза I типа ведёт дефицит одного фермента альфа-L-идуронидазы, заболевание проявляется в трёх клинических вариантах: синдром Гурлер, синдром Шейе и синдром Гурлер — Шейе.
Исторически первым двумя педиатрами: австрийским — нем. Hurler Gertrud (1889–1965) и немецким — нем. Pfaundler Meinhard von (1872–1947) описан синдром, при котором определяется подавляющее большинство компонентов фенотипа, характерных для этой группы наследственных заболеваний, и все они — резко выражены. Описанная авторами болезнь, ставшая прототипом всех мукополисахаридозов I типа, проявляется в первые месяцы жизни грубыми чертами лица (гаргоилизм), гепатоспленомегалией, тугоподвижностью суставов, деформацией позвоночного столба. Позже американским офтальмологом Шейе, англ. Н. G. Scheie (1909–1990) описана вторая форма болезни с более поздним началом и более доброкачественным течением, названная синдром Шейе. Последней описана промежуточная форма болезни, названная синдром Гурлер — Шейе.
Синдром Гурлер
Дети с синдромом Гурлер отличаются низкорослостью (отставание в физическом развитии наблюдается с конца первого года жизни). Характерны признаки гаргоилизма: крупный череп, крутой лоб, запавшая переносица, толстые губы, большой язык, характерное выражение лица («лицо выплевывающего воду»). Кроме того, отмечается короткая шея, ограниченная подвижность суставов (тугоподвижность, главным образом, затрагивает локтевые и межфаланговые суставы пальцев кистей и стоп), фиксированный кифоз на месте перехода грудных позвонков к поясничным, укорочение конечностей, в основном, происходит за счёт проксимальных отделов (бёдер и плеч), в меньшей степени — голеней и предплечий. Весьма своеобразно строение кисти пациента: пальцы короткие, одинаковые по длине (изодактилия) расходятся веерообразно, напоминая трезубец. Нижнепоясничный лордоз способствует выпячиванию живота вперёд, а ягодиц назад. Отмечается гепатоспленомегалия, склонность к формированию пупочной грыжи. Характерно диффузное помутнение роговицы за счёт накопления в ней дерматансульфата. Возможно развитие слабоумия, кариеса зубов, характерной формы ногтевых пластинок в виде часовых стёкол, тугоухость или глухота, низкий хриплый голос, гипертрихоз, сухие и жёсткие волосы. В большинстве случаев в патологический процесс вовлекается сердце — оно увеличивается в размерах, происходят изменения клапанов, миокарда, эндокарда, крупных и коронарных артерий. При рентгенологическом исследовании определяется преждевременное окостенение лямбдовидного шва, расширение турецкого седла, характерная форма позвонков («рыбьи позвонки»), искривление лучевой кости, деформации метафизарных и эпифизарных отделов длинных трубчатых костей, короткие метакарпальные кости и фаланги пальцев. Такие дети обычно не доживают до 10 лет.
Синдром Шейе
Клиническая симптоматика развивается на фоне нормальной продолжительности жизни, обычно не проявляется до достижения возраста 4 — 5 лет и может включать в себя:
- помутнение роговицы с прогрессирующей потерей зрения вплоть до слепоты;
- гаргоилизм: грубые черты лица, широкий рот с полными губами, прогнатизм;
- избыточный рост волос на теле (гипертрихоз, гирсутизм);
- характерные деформации кистей и стоп;
- тугоподвижность суставов.
Синдром Гурлер — Шейе
Синдром Гурлер — Шейе (мукополисахаридоз-I H/S) является менее тяжёлым промежуточным вариантом между синдромами Гурлер и Шейе. Фенотип пациентов также является промежуточным между фенотипом свойственным синдрому Гурлер и Шейе. Существует предположение, что больные синдромом Гурлер — Шейе являются генетическими химерами с одним аллелем синдрома Гурлер и вторым — синдрома Шейе.
Клиническая симптоматика характеризуется в основном кожными проявлениями, сочетающимися с умеренной умственной отсталостью и помутнением роговицы.
 Синдром цервикобрахиалгии
Синдром цервикобрахиалгииЛовля сига в озере
Озёрный сиг предпочитает кормиться у отмелей, и там его проще всего найти, а подходить следует с подветренной стороны. Движется в озере сиг, как правило, небольшими косяками и ловля сига на озере летом предпочтительнее на приманки имитирующие подёнку, бокоплава или нимфу. Тёмно-коричневых или серых тонов, оптимальны размер крючка для ловли озёрного сига, №3-4.
Приманку надо стараться забрасывать параллельно линии берега, и далее делать быстрые подтяжки. Делать это надо таким образом, чтобы шнур и удилище постоянно образовывали единую линию. Если приманкой служит утяжелённая нимфа, то в период проводки надо подтягивать крючок короткими рывками и делать короткие паузы. Поклёвку можно определить по короткому резкому рывку либо утяжелению шнура и сразу делать подсечку.
Диагноз
Диагноз основывается на клинических, рентгенологических и лабораторных данных. Большое диагностическое значение имеет биохимическое исследование мочи больных на кислые мукополисахариды: реакция Берри с толуидиновым синим, реакция преципитации с цетавлоном, с альбумином (см. Экспресс-методы). Отдельные фракции кислых мукополисахаридов могут быть изучены с помощью ионообменной хроматографии и электрофореза на бумаге или ацетатцеллюлозе.
При Г. выделяются в больших количествах дерматансульфат, гепарансульфат, а также хондроитинсульфаты. С возрастом экскреция с мочой дерматансульфата увеличивается.
Диагностическую ценность может иметь обнаружение мета хроматических грануляций в лейкоцитах крови и клетках костного мозга (гранулы Рейли).
Наряду с клин, и биохим, исследованием больных Г. необходимо изучение родословной и установление типа наследования, а также исследование экскреции кислых мукополисахаридов в моче родителей и родственников больных, у которых фракционный состав кислых мукополисахаридов может быть изменен.
Для обнаружения гетерозиготного носительства большое значение имеет изучение культуры фибробластов.
Рентгенодиагностика
В типичных случаях Гаргоилизм без труда распознается по рентгенограммам.
Рис. 3. Рентгенограмма таза и верхней трети бедер больного гаргоилизмом. Таз деформирован, развернут; вертлужные впадины и эпифизы бедренных костей уплощены; вальгусная деформация бедер.
Рис. 4. Рентгенограмма в боковой проекции нижних грудных и поясничных позвонков больного гаргоилизмом. Выраженный кифоз с вершиной угла на уровне L1— L2; тело L1 позвонка уменьшено в размерах, передняя поверхность террасоподобно деформирована (указано стрелкой) за счет недоразвития передне-верхнего угла.
Заболевание характеризуется генерализованным симметричным поражением эпифизов костей. Они уплощены и увеличены. Метадиафизарные отделы длинных и коротких трубчатых костей укорочены и утолщены. Типичной является вальгусная деформация проксимальных отделов бедренных костей (рис. 3). Плоские кости деформированы, утолщены, суставные впадины уплощены. Ребра также утолщены, передние концы их булавовидно расширены. Шейки ребер остаются нормальными. Межреберные промежутки сужены. Позвоночник всегда поражен, имеют место платиспондилия и угловой кифоз в нижнегрудном и верхнепоясничном отделе (рис. 4). Патогномоничным является недоразвитие передне-верхнего угла тел позвонков, обычно Th12 или L1-2. Межпозвонковые пространства не сужены. Имеется диспропорция в развитии лицевого и мозгового черепа. Основание черепа укорочено. Турецкое седло уплощено, удлинено, передние клиновидные отростки недоразвиты. Кости лицевой части черепа, особенно верхняя челюсть и носовые кости, недоразвиты. Задерживается пневматизация придаточных пазух носа. Изменяются темпы и последовательность окостенения, остеогенез происходит асимметрично.
Дифференциальный диагноз на ранних стадиях заболевания необходимо проводить с врожденной гидроцефалией (см.), рахитом (см.), гипотиреозом (см.), липоидозами (см.). Дифференциальная диагностика проводится также с другими формами хондродистрофии (см.) и эпифизарной дисплазии. При них, в отличие от Г., отмечается нормальная толщина диафизов трубчатых костей. Эпифизы имеют неправильную форму, увеличены, очертания их неровные, структура фрагментирована. Типична изодактилия. Таз деформирован, вдавлен с боков, вертлужные впадины расположены глубже, уплощены. В отличие от Г. наблюдается варусная Деформация бедренных костей, определяется системная брахиспондилия и ангуляция позвонков. При этом патогномоничной является деформация одного или двух позвонков в виде конусовидного, клиновидного заострения переднего отдела тел в нижнегрудном или верхнепоясничном отделах.
Историческая справка
Впервые заболевание, изначально названное болезнь Пфаундлера — Гурлер, описано двумя педиатрами: австрийским — (нем. Hurler Gertrud , 1889—1965) и немецким — (нем. Pfaundler Meinhard von , 1872—1947).
Описанная авторами болезнь проявляется в первые месяцы жизни грубыми чертами лица (гаргоилизм), гепатоспленомегалией, тугоподвижностью суставов, деформацией позвоночного столба. Затем американским офтальмологом Шейе, (англ. Н. G. Scheie, 1909—1990) описана вторая форма болезни с более поздним началом и более доброкачественным течением, названная синдром Шейе. Позже описана промежуточная форма болезни, названная синдром Гурлер — Шейе.
Эпоним
Заболевание названо в честь первооткрывателей — австрийского педиатраГертруды Гурлер (нем. Gertrud Hurler, 1889—1965) и американского офтальмолога Гарольда Шейе, (англ. Harold Glendon Scheie, 1909—1990).
Признаки и симптомы
Помутнение роговицы у 30-летнего мужчины с МПС VI. Синдром Гурлера и другие расстройства МПС также могут проявляться помутнением роговицы.
Дети с синдромом Гурлера могут казаться нормальными при рождении, а симптомы у них развиваются в течение первых лет жизни. Симптомы у разных пациентов различаются.
Одна из первых аномалий, которую можно обнаружить, — огрубление черт лица; эти симптомы могут начаться в возрасте 3-6 месяцев. Головка может быть большим с видными лобных костей . Череп может быть удлиненным . Нос может иметь уплощенную переносицу с непрерывными выделениями из носа. Глазницы могут быть широко расставлены, а глаза могут выступать из черепа. Губы могут быть большими, и больные дети могут постоянно держать челюсти открытыми. Скелетные аномалии возникают примерно в возрасте 6 месяцев, но могут не проявляться клинически до 10-14 месяцев. Пациенты могут испытывать изнуряющие деформации позвоночника и бедра, синдром запястного канала и жесткость суставов. Пациенты могут иметь нормальный рост в младенчестве, но перестать расти к 2 годам. Они не могут достигать высоты более 4 футов.
Другие ранние симптомы могут включать паховые и пупочные грыжи . Они могут присутствовать при рождении или развиваться в течение первых месяцев жизни. Помутнение роговицы и дегенерация сетчатки могут произойти в течение первого года жизни, что приведет к слепоте. Часто наблюдаются увеличенные печень и селезенка . Дисфункции органа нет, но отложение ГАГ в этих органах может привести к значительному увеличению размера. У пациентов также может быть диарея . Может возникнуть заболевание аортального клапана .
Обструкция дыхательных путей является частым явлением, обычно вторичным по отношению к аномальным шейным позвонкам. Часто встречаются инфекции верхних и нижних дыхательных путей.
Задержка развития может проявиться в возрасте 1-2 лет, при максимальном функциональном возрасте 2-4 года. Далее следует прогрессирующее ухудшение. У большинства детей развиваются ограниченные языковые способности. Смерть обычно наступает к 10 годам.
Ссылки
Акции и специальные предложения
Симптомы
Проявления заболевания различаются в зависимости от тяжести его течения. Различают три степени мукополисахаридоза 1 типа. При каждой разновидности симптомы могут быть более или менее выражены, ограничиваться только костными деформациями или отражаться на работе внутренних органов и нервной системы. Причем, все эти патологии с каждым годом жизни пациента все прогрессируют.
Наиболее легкая форма мукополисахаридоза 1 типа называется синдромом Шейе по имени врача, впервые описавшего патологию. Больные могут вести нормальную жизнь, у них сохраняется интеллект. Но наблюдаются патологии суставов, сердечно-сосудистой системы, нарушения зрения и слуха, характерное изменение внешности.
Промежуточное место занимает синдром Гурлера-Шейе. Он объединяет в себе признаки обоих разновидностей заболевания. Характеризуется патология нормальным интеллектом, но множественными деформациями в костной системе.

У пациентов после 2-3 лет заболевание можно обнаружить по характерным внешним признакам
Внешность пациентов
Первым симптомом заболевания является дисморфизм лица. Это огрубление черт по типу гаргоилизма начинается еще с 3-6 месяцев, а со временем только прогрессирует. Поэтому в самых тяжелых формах болезни диагноз можно поставить по фото пациента. Особенно ярко все аномалии внешности видны к 3 годам. Это, прежде всего, увеличенный череп с сильно выраженными швами и лобными буграми, короткая шея, низкий рост, деформации костей разной степени тяжести.
Черты лица ребенка также обладают характерными признаками. Они становятся к году все более грубыми. Переносица вдавленная и широкая, ноздри вывернуты, а кончик носа неестественно увеличен. Глаза широко расставлены и кажутся немного выпуклыми из-за того, что глазницы маленькие. Губы полные, язык такой большой, что не помещается во рту. Поэтому рот у больного постоянно в полуоткрытом положении. Зубы развиваются неправильно, растут нерегулярно, быстро подвергаются кариесу.
Деформации скелета
При этом заболевании почти всегда имеют место деформации скелета ребенка. Прежде всего, это видно на изменении формы грудной клетки. Ребра расширяются, и она приобретает бочкообразную форму. Примерно к году у всех детей с синдромом Гурлера развиваются дисплазии суставов и мягкие аномалии костей. Позвоночник часто искривляется, появляется кифоз, чаще всего в поясничном или грудном отделе. Позвонки выступают и приобретают характер «рыбьих». Таз недоразвит, из-за малого размера головок бедренных костей возникает дисплазия тазобедренных суставов.
Часто у пациентов развивается кистевой туннельный синдром или аномалии развития кисти по типу «когтистой лапы». Пальцы довольно короткие и плохо гнутся. Патологии мелких и крупных суставов проявляются их тугоподвижностью, частыми болями уже в раннем возрасте. Может наблюдаться вальгусная деформация стоп. У больных низкий рост, укороченная шея. Из-за неправильного телосложения они могут ходить только на цыпочках, да еще на полусогнутых ногах. Кроме того, у пациентов наблюдается мышечная гипотония и двигательная заторможенность, которая с возрастом только усиливается.
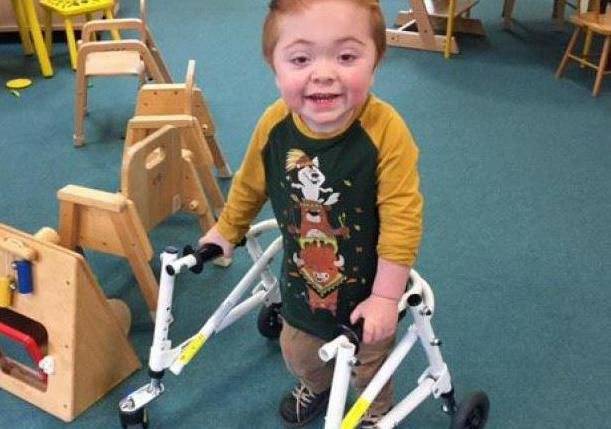
Серьезные деформации скелета, отставание в умственном и физическом развитии часто приводят к инвалидности
Внутренние органы
Дефицит фермента, ответственного за расцепление некоторых углеводов, особенно отражается на состоянии печени и селезенки. У больных уже в раннем детстве эти органы сильно увеличены, поэтому живот кажется неестественно большим. При прогрессировании заболевания может развиться цирроз печени. Страдает также сердечно-сосудистая система. Чаще всего возникает патология клапанов аорты, поэтому наблюдаются шумы в сердце. Может также развиться гипертензия, сужение коронарных сосудов или даже инфаркт.
Из-за сниженного иммунитета такие дети часто подвержены инфекционным заболеваниям. Страдают в основном органы дыхания, часто развивается дыхательная недостаточность, по ночам может наблюдаться апноэ. Характерным признаком болезни может быть также шумное дыхание пациента.
Нервная система
На втором году жизни пациента становятся заметными нарушения интеллектуального развития. Примерно до года ребенок развивается нормально, но потом начинает отставать от своих сверстников, может потерять уже приобретенные навыки. Нарушения проявляются также в речевом развитии. Это связано не только с умственной отсталостью, но и с патологиями речевого аппарата, а также с прогрессирующей глухотой.
Другие патологии
Одним из первых симптомов патологии является появление паховых и пупочных грыж у ребенка. Часто накопление мукополисахаридов в крови вызывает помутнение роговицы или развитие глаукомы. Оно может начаться еще на первом году жизни и быстро прогрессирует, приводя к слепоте. Страдает также орган слуха – у многих пациентов развивается прогрессирующая нейросенсорная тугоухость, часто возникают отиты. У больных с синдромом Гурлера меняется структура ногтей, волосы становятся жесткими, кожа – толстой и грубой.
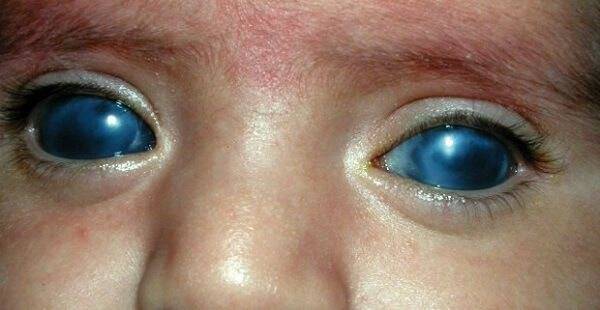
Часто у детей с синдромом Гурлера развивается помутнение роговицы или даже глаукома
Наследование
Аутосомно-рецессивный механизм наследования синдрома Гурлер — Шейе: оба родителя являются носителями дефектного гена (помечен красным кружочком). По законам Менделя 50 % детей станут носителями (как их родители), 25 % родятся генетически здоровыми и в 25 % случаев — больными.
Данная группа заболеваний наследуется, как и подавляющее большинство лизосомных болезней накопления, по аутосомно-рецессивному типу наследования. Таким образом, с одинаковой частотой встречается как у мужчин, так и у женщин.
Аутосомно-рецессивный тип наследования на практике означает, что дефектный ген расположен на одной из двух аллельных аутосом. Заболевание клинически манифестирует только в случае, когда обе аутосомы, полученные по одной от отца и матери, являются дефектными по данному гену. Как и во всех случаях аутосомно-рецессивного наследования, если оба родителя несут дефектный ген, то вероятность наследования болезни у потомства составляет 1 из 4. Таким образом в среднем, на одного больного ребёнка в такой семье приходится три без клинических признаков проявлений генной болезни. На схеме синим цветом обозначены здоровые, фиолетовым — носители дефектного гена, красным — синдром Гурлер — Шейе (два дефектных гена одной аллели). Синим кружочком помечен нормальный ген, красным — дефектный.
Лечение Мукополисахаридоза типа I-S (болезни Шейе; позднего синдрома Гурлера):
Специфическая терапия не разработана. Требуется коррекция сердечной недостаточности, которая осуществляется назначением сердечных гликозидов, диуретиков, вазодилататоров. При отсутствии эффекта от медикаментозной терапии осуществляется протезирование аортального клапана.
Клиническая картина
Синдром Гурлер — Шейе (мукополисахаридоз-I H/S) является менее тяжёлым промежуточным вариантом между синдромами Гурлер и Шейе. Фенотип пациентов также является промежуточным между фенотипом свойственным синдрому Гурлер и Шейе. Существует предположение, что больные синдромом Гурлер — Шейе являются генетическими химерами с одним аллелем синдрома Гурлер и вторым — синдрома Шейе.
Клиническая симптоматика характеризуется в основном кожными проявлениями, сочетающимися с умеренной умственной отсталостью и помутнением роговицы.
Что такое мукополисахаридоз?
Мукополисахаридозы (сокр. МПС) — это группа наследственных метаболических заболеваний, вызванных отсутствием или неисправностью определенных ферментов, необходимых организму для расщепления молекул, называемых гликозаминогликанами, — длинных цепочек сахаров (углеводов) в каждой из наших клеток. Эти клетки помогают строить кости, хрящи, сухожилия, роговицу, кожу и соединительную ткань. Гликозаминогликаны (ранее называемые мукополисахаридами) также содержатся в жидкости, которая смазывает суставы.
Больные мукополисахаридозом либо не вырабатывают достаточно одного из 11 ферментов, необходимых для расщепления этих сахарных цепей на белки и более простые молекулы, либо производят ферменты, которые не работают должным образом. Со временем эти гликозаминогликаны накапливаются в клетках крови, головном и спинном мозге, а также в соединительных тканях. Результатом является необратимое прогрессирующее повреждение клеток, которое влияет на внешний вид человека, физические способности, функционирование органов и систем и, в большинстве случаев, на умственное развитие. Симптомы могут быть похожими или различаться для разных типов расстройства.
Мукополисахаридозы классифицируются в рамках более широкой группы заболеваний, называемых лизосомными болезнями накопления. Это заболевания, при которых большое количество молекул, которые обычно распадаются на более мелкие части во внутриклеточных компартментах, называемых лизосомами, накапливаются в вредных количествах в клетках и тканях организма, особенно в лизосомах. Основная функция лизосом — переваривать нефункциональные клетки и другие материалы (включая бактерии и клеточный мусор).
Другой лизосомной болезнью накопления, которую часто путают с мукополисахаридозом, является муколипидоз. При этом заболевании в дополнение к более мелким углеводам, называемым сахарами, накапливается чрезмерное количество жировых веществ, известных как липиды (еще один основной компонент живых клеток). Больные муколипидозом могут иметь общие клинические особенности, связанные с мукополисахаридозами (определенные черты лица, аномалии костной структуры и повреждение головного мозга), однако эти болезни разные.
Патологическая анатомия
При морфологическом исследовании выявляется поражение различных органов и систем с накоплением высокомолекулярных веществ липоидно-полисахаридной природы. Эти вещества откладываются в виде мелкозернистой массы в клетках мозга, сетчатки глаз, периферических нервных ганглиев. Нервные клетки представляются набухшими, увеличенными в размерах, ядро сдвинуто к периферии. Субстанция Ниссля исчезает или уменьшается, видны лишь остатки, сдвинутые в сторону вместе с ядром. Твердая мозговая оболочка обычно утолщена. Гомогенные зернистые массы откладываются также в клетках опорной ткани, роговицы, внутренних органов (печени, селезенки, эпителия дистальных отделов извитых канальцев почек).
В клетках печени и почек наряду с кислыми мукополисахаридами откладывается значительное количество нейтральных жиров. Характерным является изменение костей — нарушение энхондрального окостенения, разрастание кровеносных сосудов в хряще и накопление в его клетках кислых мукополисахаридов и липидов. В межуточном веществе соединительной ткани выявляются только растворимые мукополисахариды, липоидные вещества не обнаруживаются. Морфологически выявляется дезорганизация структуры основного вещества, утолщение и гомогенизация коллагеновых волокон. В значительной степени страдают мозг, печень, селезенка, глаза. Наблюдаются патологические изменения в сердечных клапанах, аорте, кровеносных сосудах мозга.

 Туннельный синдром
Туннельный синдром Импиджмент-синдром плечевого сустава
Импиджмент-синдром плечевого сустава Синдром щелкающей лопатки
Синдром щелкающей лопатки Синдром карпального канала кисти
Синдром карпального канала кисти