Содержание
- 1 Классификация
- 2 Что такое амиотрофия Верднига-Гоффмана?
- 3 Симптоматика заболевания
- 4 Виды СМА
- 5 Лечение и прогноз амиотрофии Верднига-Гоффмана
- 6 Показания для применения SCS
- 7 Методы диагностики заболевания
- 8 Кипрей узколистный — фото, полезные свойства, применение в лечении, рецепты
- 9 Причины
- 10 Краткие сведения про амиотрофию Верднига-Гоффмана
- 11 Диагностика
- 12 Уход в домашних условиях
- 13 Что такое амиотрофия Верднига-Гоффмана?
- 14 Симптомы
- 15 Стимуляция спинного мозга (SCS)
Классификация
СМА проявляется в широком диапазоне степени тяжести, поражая младенцев и взрослых. Спектр заболеваний разделен на 3–5 типов в соответствии с наивысшей достигнутой вехой в моторном развитии.
Традиционная, наиболее часто используемая классификация выглядит следующим образом:
| Тип | Эпоним | Обычный возраст начала | Характеристики | OMIM |
|---|---|---|---|---|
| SMA 0 | Дородовой | Очень редкая форма, симптомы которой проявляются до рождения (снижение подвижности плода). У больных детей обычно есть только одна копия гена SMN2 , и они обычно выживают всего несколько недель даже при интенсивной респираторной поддержке. | ||
| SMA 1 (детский) | Болезнь Верднига – Гофмана | 0–6 месяцев | Тяжелая форма проявляется в первые месяцы жизни, обычно с быстрым и неожиданным началом (« синдром гибкого ребенка »). Дети никогда не учатся сидеть без опоры. Быстрая смерть мотонейронов вызывает неэффективность основных органов тела, особенно дыхательной системы. Дыхательная недостаточность, вызванная пневмонией, является наиболее частой причиной смерти. Без лечения и без респираторной поддержки дети с диагнозом СМА типа 1 обычно не доживают до двухлетнего возраста. Известно, что при надлежащей респираторной поддержке люди с более легкими фенотипами СМА 1 типа, на которые приходится около 10% случаев СМА 1, доживают до подросткового и взрослого возраста. | |
| SMA 2 (средний) | Болезнь Дубовица | 6–18 месяцев | Промежуточная форма затрагивает людей, которые были в состоянии хотя бы какое-то время в своей жизни сохранять сидячее положение, но никогда не учились ходить без опоры. Начало слабости обычно наблюдается в период между 6 и 18 месяцами жизни. Известно, что прогресс сильно различается: некоторые люди со временем постепенно ослабевают, в то время как другие при тщательном уходе остаются относительно стабильными. У этих детей обычно присутствует сколиоз, и коррекция с помощью фиксации позвоночника , растущих стержней или спондилодеза может помочь улучшить дыхание. Мышцы тела ослаблены, и респираторная система вызывает серьезную озабоченность. Ожидаемая продолжительность жизни снижается, но большинство людей со SMA 2 доживают до взрослого возраста. | |
| SMA 3 (ювенильный) | Болезнь Кугельберга-Веландера | > 12 месяцев | Юношеская форма обычно проявляется после 12 месяцев и описывает людей, которые могли ходить без поддержки хотя бы в течение некоторого времени в своей жизни, даже если позже они потеряли эту способность. Поражение дыхательных путей встречается реже, а ожидаемая продолжительность жизни нормальная или близкая к норме. Большинству людей с SMA 3 требуется поддержка мобильности. | |
| SMA 4 (начало у взрослых) | Совершеннолетие | Форма с началом у взрослых (иногда классифицируемая как СМА типа 3 с поздним началом) обычно проявляется после третьего десятилетия жизни постепенным ослаблением мышц ног, что часто требует от человека использования вспомогательных средств для ходьбы. Другие осложнения возникают редко, и это не влияет на продолжительность жизни. |
В соответствии с новыми классификациями пациенты классифицируются на «не сидящих», «сидящих» и «ходящих» на основе их фактического функционального статуса.
Двигательное развитие и прогрессирование заболевания у людей с СМА обычно оценивается с использованием проверенных функциональных шкал — CHOP-INTEND (Тест нервно-мышечных заболеваний детской больницы Филадельфии) или HINE (Хаммерсмитский неврологический осмотр новорожденных) для младенцев; и либо MFM (Измерение двигательной функции), либо один из нескольких вариантов HFMS (Функциональная двигательная шкала Хаммерсмита) у пожилых пациентов.
Одноименное название болезни Верднига – Гофмана (иногда с ошибками с одним n ) относится к самым ранним клиническим описаниям СМА у детей, сделанным Иоганном Хоффманном и Гвидо Верднигом . Одноименный термин болезнь Кугельберга-Веландера возник в честь Эрика Класа Хендрика Кугельберга (1913–1983) и Лизы Веландер (1909–2001), которые отличили СМА от мышечной дистрофии. Редко используемая болезнь Дубовица (не путать с синдромом Дубовица ) названа в честь Виктора Дубовица , английского невролога, автора нескольких исследований промежуточного фенотипа СМА.
Что такое амиотрофия Верднига-Гоффмана?
Спинальная амиотрофия 1-го типа или, по-другому, спинальная амиотрофия Верднига-Гоффмана
— это особое заболевание нервной системы, передающееся по наследству (чаще всего от обоих родителей). Эта патология характеризуется наличием мышечной слабости практически во всей мышечной системы организма. Ребёнок, страдающий от такого заболевания, не может самостоятельно сидеть, передвигаться и обслуживать себя.
К большому сожалению в мире не существует лекарства от этого типа заболевания. Максимум, что могут предложить врачи в наше время — дородовая диагностика. Такое обследование помогает избежать рождения больного малыша в семье.
Свое название патология получила от двух учёных, впервые описавших её в конце 19 в. В настоящее время под понятием спинальной амиотрофии понимается несколько форм болезни, отличающихся клинически. Но все они при этом связаны одним и тем же генетическим дефектом, которым обладают родители ребёнка.
Клиническая картина заболевания
Спинальная амиотрофия имеет несколько форм и разновидностей
 Спинальная мышечная атрофия: виды и причины
Спинальная мышечная атрофия: виды и причины, каждая из которых отличается возрастом появления характерных симптомов, тяжестью протекания заболевания и продолжительностью жизни пациентов.
Обычно эта патология приводит к инвалидности
, поскольку нарушается двигательная система организма, и пациент не способен ни самостоятельно передвигаться, ни самостоятельно себя обслуживать. При тяжелых клинических ситуациях может понадобиться постоянный врачебный контроль в повседневной жизни.
Передвигаться такому больному помогают инвалидные кресла, ходунки, костыли, трости. К смертельному исходу такое заболевание может привести только в том случае, когда появляются осложнения со стороны дыхательной и сердечно-сосудистой системы (при пневмониях и сердечной недостаточностью).
Под воздействие патологии не попадают чувствительные нервные волокна
, поэтому у ребёнка сохраняются все виды чувствительности. Не страдают также интеллект и ментальные функции, так при обучении ребёнок совершенно нормально воспринимает и усваивает информацию.
Классификация заболевания
В зависимости от возраста, при котором появились характерные симптому заболевания, амиотрофия Верднига-Гоффмана делится на несколько видов :
- Врожденная форма патологии . Примерный возраст появления изменений: от 0 до 6 месяцев. Обычно характеризуется слабым внутриутробным шевелением плода. При врождённой форме мышечная гипотония наблюдается с первых дней жизни малыша. В течение короткого времени происходит угасание глубоких рефлексов: ребёнок слабо кричит, плохо сосет молоко матери или соску, не может держать головку. Иногда случается так, что эти симптомы проявляются несколько позже, поэтому малыш может учиться держать головку и сидеть, но, поскольку имеется нарушение, эти навыки у него не разовьются. Также врожденная форма может сопровождаться бульбарными нарушениями, снижением глоточного рефлекса и фасцикулярными подергиваниями языка. Врожденная форма считается наиболее злокачественной и часто может сочетать в себе ещё и олигофрению, деформации грудной клетки, и 4 степени сколиоза . Быстрая обездвиженность и парез дыхательной системы приводит к дыхательной недостаточности и впоследствии к летальному исходу;
- Ранняя детская форма. При данной разновидности патологии первые симптомы могут проявиться после 6 месяцев. К этому моменту дети имеют нормальное физическое и психическое развитие. Они начинают потихоньку приобретать первые естественные навыки, вроде умения держать головку, стоять, садиться и переворачиваться. В большинстве случаев, при наличии данного типа заболевания, дети так и не научатся ходить. На начальной стадии возникают парезы в нижних конечностях, затем довольно быстро они развиваются в верхних конечностях и во всей мускулатуре. Наступает мышечная гипотония, угасают глубокие рефлексы, может проявиться тремор пальцев, непроизвольные мышечные сокращения. На более поздних этапах ко всем симптомам добавляются бульбарные нарушения, дыхательная недостаточность (прогрессирующая). Эта форма заболевание протекает медленнее, чем врожденный тип. Больные могут дожить вплоть до 15 лет;
- Амиотрофия Кугельберга-Веландера. Самая доброкачественная из всех форм спинальной амиотрофии. Симптомы проявляются после 2-х лет, иногда в период между 15-ю и 30-ю годами. При данной форме не встречается психической задержки развития, довольно длительное время пациенты способны двигаться самостоятельно. Многие доживают до глубокой старости на полном самообслуживании.
Симптоматика заболевания
СМА 1 и СМА 2 имеет разные симптомы и признаки.
Какие существуют отдаленные последствия черепно мозговой травмы и как максимально себя обезопасить от получения повреждения головы.
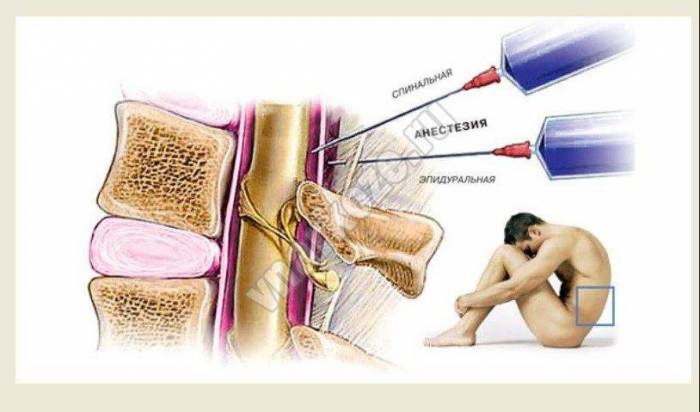 Спинальная анестезия
Спинальная анестезияРазрыв вен и сосудов головного мозга провоцирует такое заболевание, как субдуральная гематома головного мозга. В чем сложность лечения и диагностики заболевания.
Форма спинальной амиотрофии Верднига СМА 1
Первые симптомы выявляют еще во время беременности по слабому шевелению плода.
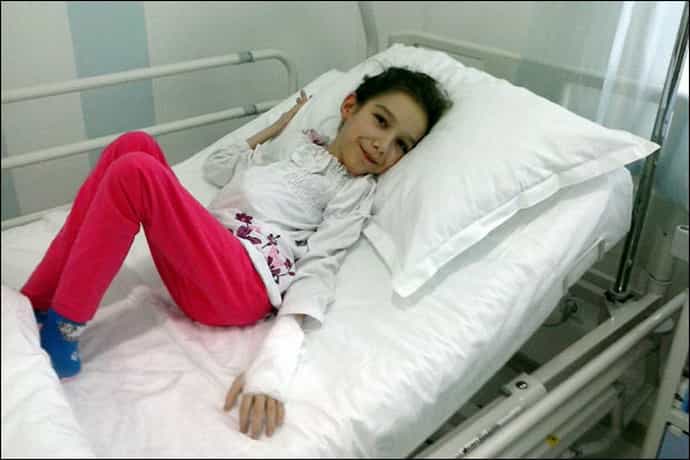
Фото: спинальная амиотрофия Верднига Гофмана
С самого рождения у детей наблюдается дыхательная недостаточность, врожденная спинальная амиотрофия Верднига Гофмана отмечаются:
- низкий мышечный тонус, ребенок не держит голову, не может перевернуться;
- отсутствие рефлексов;
- нарушения сосания, глотания, подергивание языка, пальцев, слабый плач.
Малыш принимает характерную позу «лягушки» с согнутыми в суставах руками и ногами, лежа на животе. При СМА 1 нередко отмечают частичный паралич диафрагмы – синдром Кофферата.
Виды СМА
Возраст, время проявления симптомов, особенности течения патологии, прогноз позволяют выделять несколько видов заболеваний.
СМА 0
Данная форма патологии описывается редко, часто его объединяют с первым типом СМА. Болезнь – врожденная. Характеризуется полным отсутствием движений, сухожильных рефлексов, слабостью мышц, ограниченным движением суставов коленей. С самого рождения наблюдаются дыхательные нарушения.
Часто диагноз путают с перинатальной энцефалопатией или родовыми травмами. Однако в последних двух случаях дети достаточно быстро адаптируются, их состояние становится лучше. У детей со СМА улучшения не возникают, в большинстве случаев они умирают, не дожив до месяца, от осложнений.
СМА-1
Патология первого типа имеет очень тяжелое течение. Ее называют также болезнью Верднига-Гоффмана. Диагностирован этот тип может быть от рождения до 6 месяцев. Отмечается слабость мышц, их периодическое подергивание – последнее увидеть достаточно трудно из-за достаточно большого слоя жирового слоя. Дрожь может периодически пробегать по языку малыша.
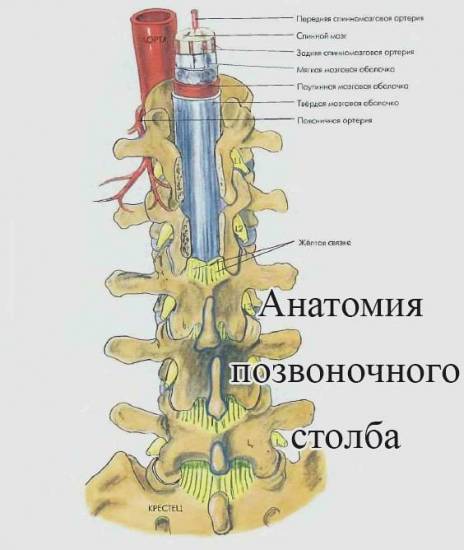 Спинальная анестезия у детей
Спинальная анестезия у детейНаблюдается ухудшение рвотного, сосательного, глотательного рефлекса, нарушение слюноотделения. Младенец не может кашлять, громко кричать. Часто сопровождается тяжелыми дыхательными нарушениями, пневмонией.
Грудная клетка у таких детей имеет более плоскую форму из-за слабо развитых мышц груди.
Малышей со спинальной амиотрофией Верднига-Гоффмана легко узнать по позе лягушонка. Бедра и плечи отведены, локти и колени согнуты.
К 6 месяцам ребенок может научиться держать головку, но практически никогда не сможет самостоятельно сесть, встать, ходить. Проблемы с глотанием вызывают сложности в кормлении.
Часто именно это заболевание сопровождается олигофренией, врожденными нарушениями работы сердца, небольшим размером головы.
Поздняя младенческая
Патология второго типа обнаруживается у малышей в возрасте от полугода до полутора-двух лет. Болезнь Дубовица характеризуется слабостью и тремором в глубоких отделах мышц, дрожью пальцев, языка, ограничением объема движения конечностей. Детей отличает маленький вес, задержка развития. Они сидят, сами кушают, но вставать и ходить не могут.
Болезнь носит прогрессирующий характер. Со временем слабеют мышцы груди, шеи, исчезают сухожильные рефлексы, отмечаются нарушения глотания, слабый голос. Больного можно узнать по свисающей головке.
Ювенильная
Патологию Кугельберга-Веландера диагностируют часто после 2 лет. Она считается относительно легкой формой СМА, многие больные доживают до 30-40 лет. Человек стоит, однако дается ему это с трудом из-за очень слабых мышц. Происходит постепенная атрофия мышц.
Ребенок до 10-12 лет развивается нормально, потом начинает спотыкаться, падает, теряет способность заниматься спортом, бегать, выходить из дома, просто перемещаться без инвалидного кресла. Больного мучают периодические судороги конечностей. Развивается сильный сколиоз, изменяется форма грудной клетки.
Часто у таких пациентов происходят переломы, отмечается ограниченный объем движения суставов.
Поздние патологии
К четвертому типу относят бульбоспинальную амиотрофию Кеннеди, дистальную амиотрофию Дюшенна-Арана, а также перонеальную амиотрофию Вюльпиана. Заболевания обычно диагностируются в возрасте 35-40 лет, иногда возрастные границы расширяются от 16 до 60 лет. Больной отмечает постепенную потерю мышечной силы, угасание рефлексов сухожилий, видимые сокращения мышц.
При атрофии Дюшенна-Арана прежде всего поражаются кисти рук. Амиотрофию Вюльпиана можно узнать по формированию крыловидных лопаток.
Лечение и прогноз амиотрофии Верднига-Гоффмана
Этиопатогенетическая терапия не разработана. В настоящее время амиотрофия Верднига-Гоффмана лечится путем улучшения метаболизма периферической нервной системы и мышечной ткани с целью замедлить прогрессирование симптомов. В терапии применяют комбинации препаратов различных фармакологических групп: нейрометаболиты (препараты на основе гидролизата головного мозга свиньи, витамины гр. В, гамма-аминомасляная кислота, пирацетам), облегчающие нервно-мышечную передачу (галантамин, сангвинарин, неостигмин, ипидакрин), улучшающие трофику миофибрилл (глютаминовая к-та, коэнзим Q10, L-карнитин, метионин), улучшающие кровообращение (никотиновая к-та, скополамин). Рекомендована лечебная физкультура и мягкий массаж.
Современное развитие техники позволило несколько облегчить жизнь пациентов и их родственников, благодаря применению автоматизированных инвалидных колясок и портативных аппаратов ИВЛ. Улучшить подвижность пациентов помогают различные методы ортопедической коррекции. Однако основные перспективы в лечении СМА связаны с развитием генетики и поисками возможностей коррекции генетических аберраций методами генной инженерии.
Врожденная амиотрофия Верднига-Гоффмана имеет крайне неблагоприятный прогноз. При ее манифестации в первые дни жизни ребенка, его гибель, как правило, происходит до 6-месячного возраста. При начале клиники после 3-х месяцев жизни, летальный исход наступает в среднем к возрасту 2 года, иногда — к 7-8 годам. Ранняя детская форма характеризуется более замедленным прогрессированием, дети погибают в возрасте 14-15 лет.
Показания для применения SCS
Эпидуральная электростимуляция спинного мозга влияет на ряд болезненных синдромов. В особенности, делать ее рекомендуется при хронической доброкачественной боли нейропатической этиологии (возникает в результате повреждения периферических нервов или корней). Полезна процедура при смешанной боли, содержащей нейропатический и ноцицепторный компонент.
Проводить SCS рекомендуется при следующих состояниях:
- синдром постдизотомии, в клинике которого преобладает болезненность в нижних конечностях;
- спастика;
- фантомная боль;
- ДЦП;
- комплексный региональный болевой синдром (ранее называвшийся альдодистрофическим синдромом) II типа; меньше подходит I тип;
- болезненный синдром после повреждения периферических нервов, сплетений;
- постгерпетическая невралгия;
- полинейропатия;
- стенокардия без возможности медикаментозного или оперативного лечения;
- ишемическая болезнь нижних конечностей III-IV степени по Фонтейну.
Методы диагностики заболевания
Когда речь заходит о диагностике такого рода заболевания, то нужно учесть, что для невролога, который будет проводить обследование, очень важен возраст появления первых симптомов у малыша.
Также важна динамика развития симптомов, данные неврологического статуса (т.е. наличие/отсутствие двигательных нарушений периферического типа на фоне нормальной чувствительности), наличие/отсутствие дополнительных врожденных аномалий и костных деформаций (склиоза, кифоза, лордоза, кривошеи).
Врожденный тип заболевания может быть выявлен неонатологом. Обследование проводится с миопатиями, мышечной дистрофией (прогрессирующей), боковым амиотрофическим склерозом, полиомиелитом, ДЦП и др. Если диагноз требует максимально точного подтверждения, то применяется также электронейромиография (исследования нервно-мышечного аппарата).
Итоговый диагноз устанавливается только после получения данных о биологии мышц и исследования генетической ситуации
Изучение ДНК-анализа позволяет генетикам выяснить гетерозиготное носительство генной аберрации (важно при планировании следующей беременности). Проводится также количественный анализ числа генов локуса СМА (позволяет высчитать наличие патологического гена у родителей
Анализ ДНК, сделанный в дородовый период, может помочь снизить вероятность рождения ребёнка с патологией Вердинга-Гоффмана. Но сложность здесь заключается в том, что для получения ДНК-материала применяются инвазивные методы пренатальной диагностики (биопсия хориона, кордоцентез, амниоцентез).
Если внутриутробно болезнь подтвердится, то это выступит показанием к искусственному прерыванию текущей беременности.
Кипрей узколистный — фото, полезные свойства, применение в лечении, рецепты
Причины
Спинальная мышечная атрофия имеет аутосомно-рецессивный характер наследования.
Спинальная мышечная атрофия связана с генетической мутацией в гене SMN1 .
Человеческая хромосома 5 содержит два почти идентичные гены на месте 5q13: а теломерные копию SMN1 и центромерный копию SMN2 . У здоровых людей ген SMN1 кодирует выживание белка мотонейрона (SMN), который, как следует из названия, играет решающую роль в выживании моторных нейронов . Ген SMN2 , с другой стороны, — из-за вариации одного нуклеотида (840.C → T) — подвергается альтернативному сплайсингу на стыке интрона 6 с экзоном 8, при этом только 10–20% транскриптов SMN2 кодируют полностью функциональная выживаемость белка мотонейрона (SMN-fl) и 80–90% транскриптов, приводящая к усеченному белковому соединению (SMNΔ7), которое быстро разрушается в клетке.
У людей, страдающих СМА, ген SMN1 мутирован таким образом, что он не может правильно кодировать белок SMN — либо из-за делеции, происходящей в экзоне 7, либо из-за других точечных мутаций (часто приводящих к функциональному преобразованию SMN1 последовательность в SMN2 ). Однако почти у всех людей есть хотя бы одна функциональная копия гена SMN2 (у большинства из них 2–4), которая все еще кодирует небольшое количество белка SMN — около 10–20% от нормального уровня, что позволяет некоторым нейронам выжить. . Однако в долгосрочной перспективе снижение доступности белка SMN приводит к постепенной гибели клеток мотонейрона в переднем роге спинного и головного мозга. Мышцы, которые зависят от этих мотонейронов для нервного ввода, теперь имеют сниженную иннервацию (также называемую денервацией ) и, следовательно, уменьшенную активность центральной нервной системы (ЦНС). Снижение передачи импульса через двигательные нейроны приводит к снижению сократительной активности денервированной мышцы. Следовательно, денервированные мышцы подвергаются прогрессирующей атрофии (истощению).
Обычно в первую очередь поражаются мышцы нижних конечностей , затем мышцы верхних конечностей, позвоночника и шеи и, в более тяжелых случаях, легочные и жевательные мышцы. Проксимальные мышцы всегда поражаются раньше и в большей степени, чем дистальные .
Тяжесть симптомов SMA в значительной степени зависит от того, насколько хорошо оставшиеся гены SMN2 могут компенсировать потерю функции SMN1 . Частично это связано с количеством копий гена SMN2 , присутствующих на хромосоме. В то время как здоровые люди несут две копии гена SMN2 , люди с СМА могут иметь от 1 до 4 (или более) из них, причем чем больше количество копий SMN2 , тем меньше тяжесть заболевания. Таким образом, у большинства детей с СМА типа I имеется одна или две копии SMN2 ; люди с SMA II и III обычно имеют не менее трех копий SMN2 ; а у людей со SMA IV обычно их не менее четырех. Однако корреляция между серьезностью симптомов и количеством копий SMN2 не является абсолютной, и, по-видимому, существуют другие факторы, влияющие на фенотип заболевания.
Спинальная мышечная атрофия наследуется по аутосомно-рецессивному типу, что означает, что дефектный ген находится на аутосоме . Для наследования заболевания необходимы две копии дефектного гена — по одной от каждого родителя: родители могут быть носителями и не затронуты лично. СМА появляется de novo (то есть без каких-либо наследственных причин) примерно в 2–4% случаев.
Спинальная мышечная атрофия поражает людей всех этнических групп, в отличие от других хорошо известных аутосомно-рецессивных заболеваний, таких как серповидно-клеточная анемия и муковисцидоз , которые имеют значительные различия в частоте встречаемости среди этнических групп. Общая распространенность SMA всех типов и среди всех этнических групп находится в диапазоне 1 на 10 000 человек; частота гена составляет примерно 1: 100, следовательно, примерно каждый 50 человек является носителем. Нет никаких известных последствий для здоровья носительства. Человек может узнать о своем статусе носителя только в том случае, если его ребенок страдает СМА или после секвенирования гена SMN1 .
Больные братья и сестры обычно имеют очень похожую форму СМА. Тем не менее, случаи появления разных типов SMA среди братьев и сестер действительно существуют — хотя и редко, эти случаи могут быть связаны с дополнительными делециями de novo гена SMN , не связанными с геном NAIP , или различиями в количестве копий SMN2 .
Краткие сведения про амиотрофию Верднига-Гоффмана
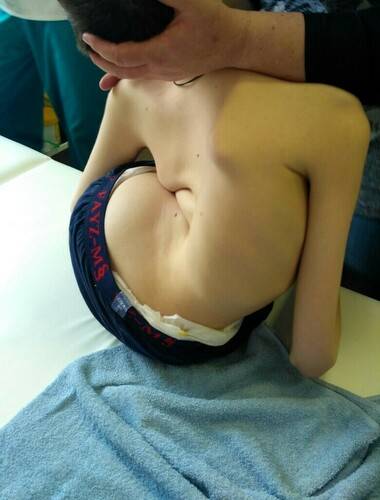 Согласно проведенным исследованиям, медики пришли к выводу, что амиотрофия Верднига-Гоффмана — наиболее серьезное заболевание среди всех существующих СМА. Заражение нового организма данным недугом составляет 1 к 6-10 тысячам появившихся на свет детей, а переносчиком неблагоприятного гена — является каждый 50-й человек. Однако, благодаря определенному типу наследования, анормальность в теле малыша может проявиться только в том случае, если подобная мутация присутствует у обоих родителей. Учитывая информацию среднестатистической проверки в подобной ситуации, риск рождения ребенка с патологией достигает 25%. Доктора разделяют амиотрофия Верднига-Гоффмана на несколько классов: врожденный, промежуточный и поздний. Последний вариант отклонения, выделяется в качестве самостоятельной болезни под названием амиотрофия Кугельберга-Веландера.
Согласно проведенным исследованиям, медики пришли к выводу, что амиотрофия Верднига-Гоффмана — наиболее серьезное заболевание среди всех существующих СМА. Заражение нового организма данным недугом составляет 1 к 6-10 тысячам появившихся на свет детей, а переносчиком неблагоприятного гена — является каждый 50-й человек. Однако, благодаря определенному типу наследования, анормальность в теле малыша может проявиться только в том случае, если подобная мутация присутствует у обоих родителей. Учитывая информацию среднестатистической проверки в подобной ситуации, риск рождения ребенка с патологией достигает 25%. Доктора разделяют амиотрофия Верднига-Гоффмана на несколько классов: врожденный, промежуточный и поздний. Последний вариант отклонения, выделяется в качестве самостоятельной болезни под названием амиотрофия Кугельберга-Веландера.
Диагностика
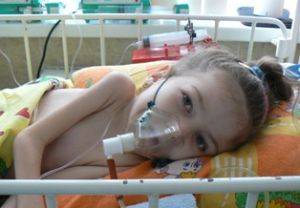 При спинальной амиотрофии Вердника диагностика заключается в проведении генетического анализа, выявляя мутации или делецию гена SMN.
При спинальной амиотрофии Вердника диагностика заключается в проведении генетического анализа, выявляя мутации или делецию гена SMN.
При обнаружении делеции теломерной копии SMNt диагноз считают подтвержденным.
В случае отсутствия делеции проводят дополнительные исследования:
- электонейромиографию;
- исследование нервной проводимости;
- тест на креатинкиназу;
- биопсию мышц и нервной ткани.
При нормальных показателях фермента креатинкиназы проводят подсчет копий SMNc. В случае единственной копии идентифицируют точечную мутацию, принимая окончательное решение.
Дифференциальная диагностика
Похожие симптомы наблюдаются при врожденной миопатии – нарушении тонуса мышц.
Полностью исключить мышечную гипотонию позволяют результаты биопсии.
Определенное сходство с заболеванием Верднига-Гофмана имеет острый полиомиелит. Он начинается бурно, с резкого подъема температуры, несимметричных множественных параличей.
Несколько дней длится острый период, затем процесс переходит в восстановительную стадию.
Гликогенозы и врожденные миопатии также характеризуются сниженным мышечным тонусом. Изменения вызываются, в отличие от спинальной мышечной амиотрофии, нарушением обмена веществ, карциномой, гормональным дисбалансом. Следует исключить также болезнь Гоше, синдром Дауна, ботулизм.
Уход в домашних условиях
Что такое амиотрофия Верднига-Гоффмана?
Спинальная амиотрофия 1-го типа или, по-другому, спинальная амиотрофия Верднига-Гоффмана — это особое заболевание нервной системы, передающееся по наследству (чаще всего от обоих родителей). Эта патология характеризуется наличием мышечной слабости практически во всей мышечной системы организма. Ребёнок, страдающий от такого заболевания, не может самостоятельно сидеть, передвигаться и обслуживать себя.
К большому сожалению в мире не существует лекарства от этого типа заболевания. Максимум, что могут предложить врачи в наше время — дородовая диагностика. Такое обследование помогает избежать рождения больного малыша в семье.
Свое название патология получила от двух учёных, впервые описавших её в конце 19 в. В настоящее время под понятием спинальной амиотрофии понимается несколько форм болезни, отличающихся клинически. Но все они при этом связаны одним и тем же генетическим дефектом, которым обладают родители ребёнка.
Клиническая картина заболевания
Спинальная амиотрофия имеет несколько форм и разновидностей, каждая из которых отличается возрастом появления характерных симптомов, тяжестью протекания заболевания и продолжительностью жизни пациентов.
Обычно эта патология приводит к инвалидности, поскольку нарушается двигательная система организма, и пациент не способен ни самостоятельно передвигаться, ни самостоятельно себя обслуживать. При тяжелых клинических ситуациях может понадобиться постоянный врачебный контроль в повседневной жизни.

Передвигаться такому больному помогают инвалидные кресла, ходунки, костыли, трости. К смертельному исходу такое заболевание может привести только в том случае, когда появляются осложнения со стороны дыхательной и сердечно-сосудистой системы (при пневмониях и сердечной недостаточностью).
Под воздействие патологии не попадают чувствительные нервные волокна, поэтому у ребёнка сохраняются все виды чувствительности. Не страдают также интеллект и ментальные функции, так при обучении ребёнок совершенно нормально воспринимает и усваивает информацию.
Классификация заболевания
В зависимости от возраста, при котором появились характерные симптому заболевания, амиотрофия Верднига-Гоффмана делится на несколько видов:
- Врожденная форма патологии. Примерный возраст появления изменений: от 0 до 6 месяцев. Обычно характеризуется слабым внутриутробным шевелением плода. При врождённой форме мышечная гипотония наблюдается с первых дней жизни малыша. В течение короткого времени происходит угасание глубоких рефлексов: ребёнок слабо кричит, плохо сосет молоко матери или соску, не может держать головку. Иногда случается так, что эти симптомы проявляются несколько позже, поэтому малыш может учиться держать головку и сидеть, но, поскольку имеется нарушение, эти навыки у него не разовьются. Также врожденная форма может сопровождаться бульбарными нарушениями, снижением глоточного рефлекса и фасцикулярными подергиваниями языка. Врожденная форма считается наиболее злокачественной и часто может сочетать в себе ещё и олигофрению, деформации грудной клетки, и 4 степени сколиоза. Быстрая обездвиженность и парез дыхательной системы приводит к дыхательной недостаточности и впоследствии к летальному исходу;
- Ранняя детская форма. При данной разновидности патологии первые симптомы могут проявиться после 6 месяцев. К этому моменту дети имеют нормальное физическое и психическое развитие. Они начинают потихоньку приобретать первые естественные навыки, вроде умения держать головку, стоять, садиться и переворачиваться. В большинстве случаев, при наличии данного типа заболевания, дети так и не научатся ходить. На начальной стадии возникают парезы в нижних конечностях, затем довольно быстро они развиваются в верхних конечностях и во всей мускулатуре. Наступает мышечная гипотония, угасают глубокие рефлексы, может проявиться тремор пальцев, непроизвольные мышечные сокращения. На более поздних этапах ко всем симптомам добавляются бульбарные нарушения, дыхательная недостаточность (прогрессирующая). Эта форма заболевание протекает медленнее, чем врожденный тип. Больные могут дожить вплоть до 15 лет;
- Амиотрофия Кугельберга-Веландера. Самая доброкачественная из всех форм спинальной амиотрофии. Симптомы проявляются после 2-х лет, иногда в период между 15-ю и 30-ю годами. При данной форме не встречается психической задержки развития, довольно длительное время пациенты способны двигаться самостоятельно. Многие доживают до глубокой старости на полном самообслуживании.
Симптомы
Существует четыре вида спинальной амиотрофии, они отличаются симптомами и длительностью жизни больного. Все формы патологии проявляются общим признаком, это нарушение умственных функций и чувствительности.
В области тазовых органов не происходят изменения. Все признаки проявляются с нарушением двигательной системы.
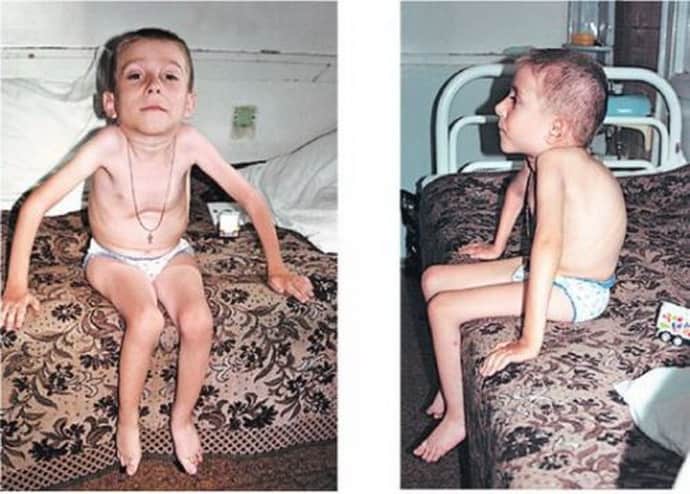
Спинальная амиотрафия 1 типа
Происходит нарушение глотательной и сосательной функции. Ребенку становится трудно двигать языком и заметно, как возникает волнообразное сокращение на нем. Крики малыша слышно слабо. Если снижаются глотательные функции, то будут проблемы с питанием, так как еда будет попадать в дыхательную систему. Как правило, это приводит к тому, что развивается аспирационная пневмония и малыш из-за этого может умереть.
Если происходит поражение межреберных мышц, то будут нарушения дыхательной системы. По началу это может быть не так заметно, но со временем состояние ребенка будет ухудшаться. Как правило, мышца лица, которые отвечает на движение глаз не нарушаются. Ребенок не сидит, не может держать и поворачивать голову и тянуться за игрушками. Если были какие-то движения развиты до спинальной амиотрофии, то они пропадут.
Кроме этих нарушений возникает и деформация грудного отдела. Если заболевание заметно стало сразу же после того, как родился ребенок, то он живет не больше полугода. Если же развитие патологии было после трех месяцев, то малыш проживет примерно два года. Летальный исход может быть куда раньше, даже из-за инфекционного поражения дыхательной системы. Спинальная амиотрофия Верднига может быть вместе с другими врожденными патологиями.

Спинальная амиотрафия 2 типа
Заболевание у малыша развивается от полугода и до двух лет. До этого момента не заметно никаких изменений. Ребенок самостоятельно держит голову, сидит, переворачивается и даже ходит.
Как при первом типе спинальной амиотрофии не происходит поражение мышц глаз и мимических. Может наблюдаться дрожь в кистях, подёргивание языка, рук и ног. Дальше развивается мышечная слабость в шее и это приводит к тому, что голова свисает. Может также быть деформация грудного отдела, сколиоз. Форма заболевания является доброкачественной и чаще всего могут быть нарушение дыхательной системы в подростковом возрасте.

Спинальная амиотрофия 3 типа
Часто болезнь встречается у больных в возрасте от двух и до пятнадцати лет. Симптомы проявляются в виде неправильной походки и мышечной слабости в конечностях.
Позже может происходить и поражение верхних конечностей. Тогда возникает нарушение мимических мышц, а глазами пациент двигает без проблем. Пропадает рефлекс тех мышц, которые уже поражены. Может возникать деформация скелета и суставов. При таком заболевании, если будет необходимое лечение больной доживает примерно до сорока лет.
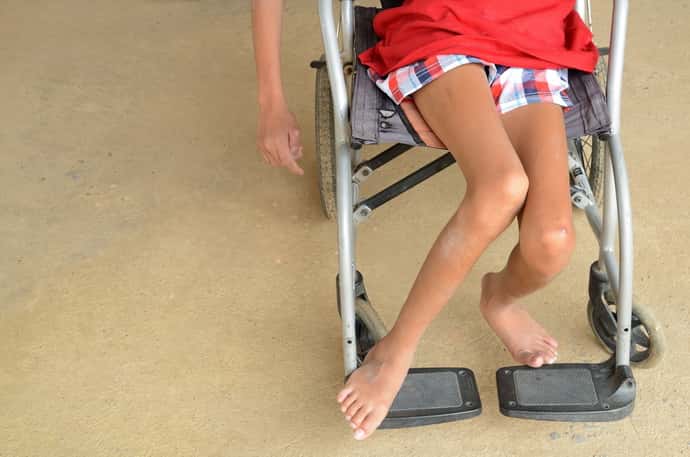
Спинальная амиотрофия 4 типа
Данный вид спинальной амиотрофии происходит уже во взрослом возрасте после 35 лет. Проявляется патология в виде мышечной слабости в конечностях и снижения рефлексов.
Стимуляция спинного мозга (SCS)
SCS осуществляется путем имплантации электродов в ЭП (эпидуральное пространство). Спинной мозг окружают три слоя оболочек, самая плотная из них – наружная. С ее помощью вокруг мозга образуется «мешок», заполненный ликвором (спинномозговая жидкость).
 Электрод устанавливают между костными стенками позвоночника и плотной наружной мозговой оболочкой. Электрод соединяют с прибором (нейростимулятором), имплантируемым подкожно.
Электрод устанавливают между костными стенками позвоночника и плотной наружной мозговой оболочкой. Электрод соединяют с прибором (нейростимулятором), имплантируемым подкожно.
Показаниями для нейростимуляции спинного мозга являются:
- Осложнения после травм позвоночника.
- Двигательные и функциональные расстройства.
- Хронические боли ног, рук, спины, шеи (невропатического характера).
- ДЦП.
- Спастика (постоянное напряжение любой группы мышц).
- Фантомные боли.
- Стенокардия (при невозможности медикаментозного лечения).
- Ишемическое заболевание нижних конечностей (третьей и четвертой степеней).
- Боли после повреждения сплетений или периферических нервов.
Существуют и противопоказания к использованию стимулятора:
- Местная или общая инфекция.
- Тяжелые дегенеративные заболевания позвоночника, вызывающие проблемы с вводом электродов.
- Патологии иммунной системы.
- Имплантация кардиостимулятора.
Перед установкой постоянного нейростимулятора в обязательном порядке проводится тестовая (временная) стимуляция. В это время и врач, и пациент наблюдают за эффективностью работы прибора при помощи наружных тестовых электродов. Обычно этот период длится несколько дней.
 Спинальная анестезия при кесаревом сечении
Спинальная анестезия при кесаревом сечении Спинномозговая (спинальная) травма
Спинномозговая (спинальная) травма Эпидуральная и спинальная анестезия при кесаревом: в чем отличие?
Эпидуральная и спинальная анестезия при кесаревом: в чем отличие? Спинальная анестезия: отзывы, особенности, возможные последствия
Спинальная анестезия: отзывы, особенности, возможные последствия